Chapter 43
Cognitive Development and Aging
The human brain has evolved over a very extensive period (see Chapter 42), but an individual human brain develops rapidly over the first few years of life, allowing the nearly helpless human infant to gain control of locomotion, language, and thought. The story of brain development in the early years of infancy through adolescence occupies the first part of this chapter. This is followed by consideration of the cognitive changes that occur during this period of the life span and how these capacities are altered over the course of normal aging. The final parts of the chapter discuss pathological processes that affect cognitive development in childhood and adolescence and can cause devastating cognitive impairment in the elderly.
Brain Development
Basic Concepts of Brain Development
Brain Maturation Progresses Well into Adolescence
Brain development is an organized, predetermined, and highly dynamic multistep process that continues beyond birth into the postnatal period, well into adolescence in humans. Most of our knowledge about human brain development has been obtained through analyses of postmortem specimens and, more recently, powerful neuroimaging techniques. These approaches have not only provided extensive information on how the size and shape of different brain regions change from conception through adulthood in relation to normal cognitive development (Fig. 43.1) but they also have opened unparalleled opportunities for identifying abnormal patterns of development in various psychiatric disorders. The brain grows rapidly in the first 2 years of life, by which time it has achieved 80% of its adult weight. Thereafter, brain growth is slower, reaching over 90% of average adult weight by the sixth year. The development of the cerebral cortical surface proceeds gradually from the flat (lissencephalic) brain of the fetus to the adult gyral pattern at birth. Cortical development in most mammals is broadly divided into two phases. The first is a genetically determined sequence of events occurring in utero that can be modified by both the local environment within the fetal nervous system and the maternal environment. The second phase is one that occurs in the perinatal period when the connectivity of the cortex becomes sensitive to patterns of neural activity. Glial processes also critically participate in normal development, supporting cell proliferation, differentiation and guidance, and the formation of myelin sheets around axons. Brain structure size, and developmental trajectories are highly variable and sexually dimorphic.

Figure 43.1 The size and form of the human brain as it develops through gestation and early infancy.
The Cortex Thickens during Development
Development of the cortical mantle in the human brain was documented extensively in the classic studies of Conel (1939–1963). Longitudinal neuroimaging studies in large samples of children have shown changes in volume of cortical gray matter that are nonlinear and regionally specific (Lenroot & Giedd, 2006). Volume of gray matter increases during preadolescence, with a maximum occurring around 10–12 years of age in the parietal lobe, 11–12 years of age in the frontal lobe, and 16–17 years of age in the temporal lobe, followed by a decline in postadolescence. Although neuroimaging techniques lack the resolution to identify the specific structural modifications that underlie changes in cortical volume during development (i.e., cell number, synapse density, myelination), they do indicate that development of the human cerebral cortex is highly dynamic over time and varies across different cortical areas. Developmental trajectories during childhood and adolescence also differ between male and female brains (Lenroot et al., 2007), suggesting that sex steroids play a significant role (Raznahan, Lee et al., 2010).
Dendritic Spine Number Increases During Development
The formation of dendritic spines and the time course of changes in the length and branching patterns of dendrites have been described for visual and frontal cortical areas in humans. Within the visual cortex, peak spine density is achieved around 5 months of age; this number then decreases until adult values are obtained around 21 months of age. Progressive elongation of dendrites occurs up to 24 months. Dendritic development in the visual system reaches mature levels in deep cortical layers earlier than in superficial layers, displaying the “inside-out” pattern of development that is characteristic of neurogenesis and migration (Chapter 15).
Development and maturation of the frontal cortex proceed more slowly. Whereas neuronal density in the primary visual cortex (V1) reaches adult levels by 4–5 months, neuronal density in the frontal cortex fails to reach adult levels even at 7 years of age. Additionally, by 2 years of age, dendritic length in frontal cortex (which is mature by 18–24 months in V1) is only half that found in adults. Left–right asymmetries exist in the dendritic branching patterns of pyramidal neurons within layer V of the inferior frontal and anterior precentral cortex. During the first year, growth is more advanced on the right side, but by 6–8 years of age, the maturation of distal dendrites on the left exceeds that of the right.
Synapses First Increase and Then Decrease in Number
As in the brains of other animals (Chapter 18), the immature human brain contains many more synapses than the mature brain (Huttenlocher, 1990). Within the primary visual cortex, synaptic density increases gradually during late gestation and early postnatal life, and then doubles from 2 to 4 months of age. After age 1, however, there is a decline in synaptic density until adult values (50 to 60% of the maximum) are attained at about 11 years. The time course of the decrease in synaptic density varies across different cortical layers (Fig. 43.2). The decrease does not display the “inside-out” pattern of development; rather, there is a considerable decrease, over time, in the number of synapses in every layer. A postnatal increase and subsequent decline in synaptic density also occurs in the middle frontal gyrus (layer III), but these frontal cortex changes take place over a longer time course than in the primary visual cortex (Fig. 43.3). The maximum density of synapses occurs around 1 year of age (compared to 4 months in the visual cortex), and adult values are not reached until roughly 16 years of age (compared to 7–11 years for the visual cortex).
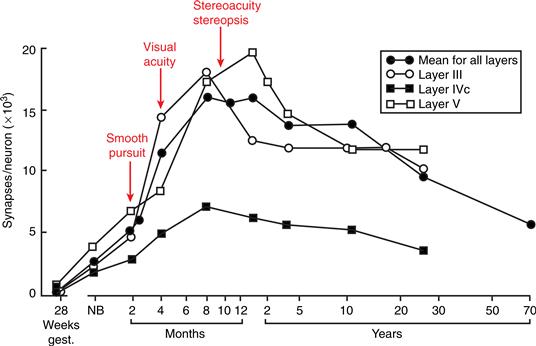
Figure 43.2 Variations in the density of synapses of different cortical layers of the human primary visual cortex during development. Arrows point to the emergence of various visual functions in relation to the increase in synapses in the visual cortex. NB, newborn. Adapted with permission from Huttenlocher and De Courten (1987).
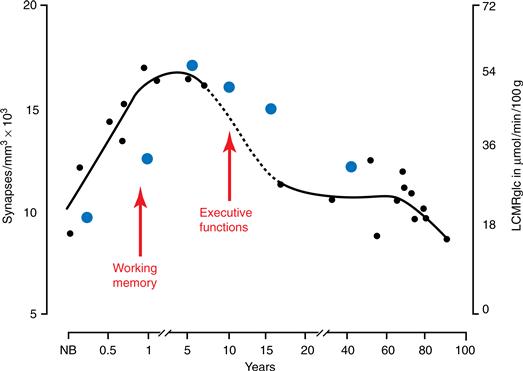
Figure 43.3 Variations in the human prefrontal cortex during development: Density of synapses in layer III of the medial frontal gyrus (black circles) and resting glucose uptake (LCMRglc) in the frontal cortex (blue circles). Arrows point to the approximate periods of emergence of various prefrontal cortex functions. NB, newborn. Adapted with permission from Huttenlocher (1990) and Chugani, Phelps, and Mazziotta (1993).
In summary, these quantitative anatomical results suggest that, in contrast to other animals (Chapter 18), programmed cell death plays only a limited role in human brain development. Synapse elimination in humans and other primates exceeds that observed in many species, however, suggesting that modifications in synaptic connectivity may be particularly important for the development of complex nervous systems. Current findings are consistent with a role for incoming afferent input in selectively stabilizing functional synapses and in eliminating or suppressing inactive contacts. The exuberant connectivity observed during development may also support compensatory processes in cases of early brain trauma, allowing surviving synapses to assume functions normally subserved by damaged inputs.
Postnatal Refinement of Synaptic Contacts Occurs in the Neostriatum and Hippocampus as Well
Although the morphological development of certain subcortical structures, such as the thalamus and cerebellum, is nearly complete at birth, other structures continue to mature into early postnatal life. In the neostriatum, synaptic density increases until the end of the first postnatal month, and changes in neuronal and neuropil morphology are observed until 2–4 months postnatally. Refinement of synaptic contacts proceeds until the end of the first postnatal year when the striatal neurochemical mosaic attains adult characteristics. In the hippocampus, neurons have reached their destination at birth in all CA fields, but immature cells continue to accrue within the dentate gyrus throughout the first year of life. In addition, continued dendritic remodeling in the hippocampus has been observed as late as the 5th postnatal year in humans (Seress, 2008). Myelination in the subicular and presubicular regions, a key relay zone between the hippocampus and many cortical areas, continues through adolescence and adulthood (Benes, Turtle, Khan, & Farol, 1994) with female subjects showing a greater extent of myelination between the ages of 6 and 29 than males. These changes are accompanied by corresponding changes in the volume of the hippocampus during early development (Giedd et al., 1996).
White Matter Increases Throughout Childhood and Adolescence
In contrast to the inverted U shape of gray matter development, white matter maturation in the brain continues long after the gray matter has reached its definitive volume (Lenroot & Giedd, 2006; Klingberg, 2008). In general, myelination progresses from caudal to cephalic. Thus, the brainstem myelinates prior to the cerebellum and basal ganglia. Similarly, the cerebellum and basal ganglia myelinate prior to the cerebral hemispheres. Sensory and motor systems display mature myelination within the first 2 years of life. However, nonspecific thalamic radiations do not reach mature levels until 5–7 years of age, and intracortical fibers continue their myelination processes well into the third decade of life. Numerous morphological studies demonstrate that the corpus callosum is uniformly thin in the first month postnatally. The genu and then the splenium undergo rapid growth spurts during the next 5 months, resulting in their characteristic “bulbous” appearance.
Cerebral Metabolic Rate Increases and then Decreases after Birth
Increases in neuronal activity have been linked to increases in cerebral metabolism, as measured by neuroimaging techniques. Data show substantial subcortical activation in newborns, notably in the thalamus and phylogenetically old portions of the cerebellum, but little activation of the cerebral cortex. However, over the first 3–4 years of life, cortical metabolic rate increases until it reaches levels twice those observed in adults. After 4 years of age, metabolic activity declines gradually to adult levels at around age 15. The time course of the rise and decline of activity, as revealed by positron emission tomography (PET), varies across cortical regions but parallels the rise and decline in the number of synapses in these areas, suggesting that the exuberant cortical synapses observed during development are metabolically active. As these synapses are eliminated, metabolic activity decreases.
Dorsolateral Prefrontal Cortex Is One of the Last Brain Regions to Develop
Refinement in the circuit organization of the dorsolateral prefrontal cortex, like that of the hippocampal formation (see earlier discussion), continues until quite late in development. In the case of the dorsolateral prefrontal cortex, a gradual decline in synaptic density continues well into late adolescence and suggests that connectivity in this cortical region undergoes substantial change until 15–20 years of age. These changes are accompanied by a decrease in metabolic rate during the same period (Fig. 43.3). It is believed that changes in the neuromodulatory effects of dopamine may influence the adolescent refinement of excitatory and inhibitory inputs to layer III pyramidal neurons and that dopamine may have a particularly strong influence on cortical information processing around the time of puberty (for the relevance of this late prefrontal cortex development in the emergence of psychopathologic states, such as schizophrenia, see the section on pathological processes).
Summary
Structural, metabolic, and physiological indices of human brain development all point to a long postnatal time course that displays considerable variability from region to region and from system to system. Thus, while postnatal development in the hippocampus proceeds until approximately 4–5 years of age, it continues until 7–11 years for the visual cortex and until around 16 years of age for the frontal cortical areas. The dorsolateral portion of the prefrontal cortex appears to be one of the last regions of the brain to develop. Interestingly, the development of myelin has been observed as late as the third to fourth decade of life for fibers within the association areas of the neocortex. Finally, brain structure, size and developmental trajectories are sexually dimorphic.
Cognitive Development and Aging: A Life Span Perspective
Investigators are now attempting to link the types of neural changes described in the preceding section to the maturation of sensory, behavioral, and cognitive capacities in infants and children. Such studies are limited in number. Until recently, the predominant view was that postnatal development of the cortex is largely intrinsically determined. However, new studies of both animals and humans have revealed a central role for extrinsic factors in shaping the organization of neural systems and in permitting recovery from brain damage. Indeed, calculations showing that information in the genome is not sufficient to specify the connectivity of the brain, together with evidence for the long-lasting existence of transient, redundant connections in primates, suggest that neural changes under the influence of environmental input play a significant and persistent role in the development of functional specificity in the human brain. Thus, genetic and environmental influences are intimately intertwined in guiding brain development (Lenroot et al., 2009).
Cognitive Development in the First Years of Life
Anatomical Development of the Visual System in Infants Is Linked to Functional Development
Several parallels have been noted between anatomical changes and the emergence of visual system function in humans. The visual abilities of the newborn have been linked to subcortical structures that show mature anatomy and high metabolic rates during this period. The limited visual abilities of the newborn are augmented by the appearance of smooth pursuit tracking around 6–8 weeks. The emergence of this capacity may be related to the maturation of cortical layers IV, V, and VI in V1 (Fig. 43.2). These layers connect magnocellular afferents from the lateral geniculate nucleus and the middle temporal (MT) region, a pathway important for the detection of visual movement. Visual acuity and visual alertness increase dramatically around 4 months of age, when the volume of visual cortex reaches adult levels and the highest density of synapses is present. Shortly thereafter, cortically mediated binocular interactions become apparent. These interactions, which include stereoacuity, binocular summation of the light reflex, and stereopsis (binocular depth perception), appear in the same time frame as maturation of the middle cortical layers and rapid synaptogenesis in V1. This period of rapid growth appears to be a time of increased vulnerability to altered afferent input. Strabismic amblyopia and amblyopia due to the absence of patterned input (i.e., centrally mediated visual impairments) are reported to occur at this age unless corrected early. The time period during which visual impairments can be corrected is different for different visual functions. For example, correction for an absent lens (aphakia) due to cataracts may be completely effective only when performed prior to 2 months of age—that is, just prior to the onset of exuberant synaptogenesis in V1. However, very high synaptic density persists to at least the age of 4 years in V1. The presence of this extensive connectivity may account for the ability to recover from amblyopia (with forced use of the strabismic eye) during this time. If the disruption of binocular convergence by strabismus is not corrected within the first year of life, the ability to see stereoscopically may not develop even though the development of acuity and contrast sensitivity is normal. Thus, there may be separate critical periods (see Chapter 21) for the development of resolution acuity and stereopsis. Because the parvocellular system is thought to underlie acuity, whereas disparity detection is mediated by the magnocellular system, the magnocellular system may be more modifiable by environmental input than the parvocellular system. This differential sensitivity may be due to the slower maturation of the magnocellular system and/or to differences in the number of exuberant synapses within these systems.
Studies of individuals born deaf and blind suggest that in humans, as in other animals, there is a time period when cortical areas that normally process information from the deprived modality can be reorganized to process information from intact modalities. From the alteration of visual functions seen in individuals born deaf, it appears that areas of the auditory cortex have been recruited for visual function. Deaf subjects, by comparison, show abnormally strong electrical responses to peripheral visual stimuli. These responses are not found in normally hearing individuals, even those exposed to sign language by deaf parents. The prolonged persistence of exuberant cortico-cortico connections may provide the substrate for such cortical reorganization.
Development of the Ventral Visual Stream and Object Representation
A multitude of visual cortical areas located in the striate and extrastriate cortex contribute to different aspects of visual processing (see Chapter 44). These visual areas have been divided into two visual processing systems, one coursing ventrally from the striate and extrastriate areas to the ventrolateral temporal cortex and the other coursing dorsally from the striate and extrastriate cortex to the parietal cortex. The dorsal stream mediates spatial processing associated with attention to movement and location, whereas the ventral stream is involved primarily in processing information about patterns and objects. Both streams project rostrally, each reaching common and adjacent areas of the prefrontal cortex.
Major functions of the ventral stream include the ability both to segment a pattern into a set of constituent parts (local level of processing) and to integrate those parts into a coherent whole (global level of processing). In adults, systematic differences exist in the distribution of global and local levels of processing within the brain, such that global levels of processing are associated with the right posterior temporal region and local levels of processing with the left posterior temporal regions. Around 4 months of age, infants show hemispheric differences for global and local processing that are similar to adults. Thus, at this age, children segment out well-formed units and use simple associations to organize these units into a configuration. With further maturation, children show changes in the way they decompose objects into parts and in the relations they use to organize the parts. These changes may reflect the protracted development of the posterior temporal cortical areas, a finding consistent with ERP studies in human infants indicating functional activation within the temporal cortical areas around 6 months of age. The latter observation parallels the emergence, around the same age, of object representation and visual rule learning (Alvarado & Bachevalier, 2000).
Development of the Dorsal Visual Stream and Spatial Attention
A variety of spatial processes have been associated with activation of the dorsal visual pathway, such as the processing of information about spatial attention and spatial location (see Chapter 45). Considerable clinical and experimental evidence shows that the posterior parietal lobe plays a crucial role in the ability to shift attention to different spatial locations. By at least 6 months of age, infants, like adults, show facilitated stimulus detection as a function of prior cueing—that is, they take less time to detect an object if it is presented at a location that has been cued previously. They also demonstrate, as do adults, quicker target detection when the interval between the cue and target is short than when this interval is long. While this basic attentional response may be robust as early as 4 months of age, the timing parameters that elicit the response change with development, suggesting that further maturational processes occur in the parietal cortex.
Precursors of Declarative Memory Emerge Early in Development
Looking preference has been used to study the emergence of recognition memory ability in both human infants and monkeys (Bachevalier & Vargha-Khadem, 2005). In both species, the most common procedure is to first present the infant with an attractive stimulus for a period of time (generally 20 to 30 s). After an intervening delay that can vary from a few seconds to many hours, the familiar stimulus is shown together with a new item. Human infants as young as 1 day old display a strong preference for looking at the novel object after delays as long as 24 h, demonstrating that they recognize the previously presented item as familiar. The early emergence of recognition memory is also observed in infant monkeys and, in this species, surgical removal of the hippocampal formation and adjacent tissue before the age of 3 weeks disrupts this ability only when the hippocampus normally becomes fully mature (around 1 year of age in monkeys). Thus, early developing recognition memory processes are likely mediated by allocortical areas (phylogenetically older cortex) of the medial temporal lobe, such as the perirhinal cortex and parahippocampal gyrus. These cortical areas are known to develop earlier than the hippocampus (Zeamer & Bachevalier, 2010). The capacity for recall over long periods of time, as measured by nonverbal deferred imitation tasks, also emerges by the end of first year in humans and is impaired after early hippocampal injury (Bauer, 2002; de Haan, Mishkin, Baldeweg, & Vargha-Khadem, 2006).
The early emergence and continued improvement of recognition and recall abilities is consistent with what is known about maturation of the hippocampal formation in both humans and monkeys (see earlier discussion). In both species, the hippocampal formation is almost adult-like at birth, although there are a number of postnatal morphological refinements that continue until the end of the first year in monkeys and until about 4–5 years of age in humans. These protracted modifications in hippocampal circuitry, together with the further maturation of cortical areas in the temporal and prefrontal lobes, may provide a basis for the similarly late elaboration of certain capacities, including spatial and relational memory (see Chapter 48). Evidence that contributions of the medial temporal lobe and prefrontal cortex to declarative memory follow distinct developmental trajectories has also been documented by functional neuroimaging (Ofen et al., 2007).
Orientation to Faces, Recognition of Mother’s Face, and Imitation of Facial Expressions Are Present at Birth
The human face is a powerful visual source of social information, and the development of face-processing abilities appears to follow a delayed postnatal development. Newborns preferentially orient to faces, recognize the mother and other familiar faces, and imitate facial expressions. These precocious tendencies appear to be mediated by a subcortical retinotectal pathway. For example, newborns display preferences selectively for moving stimuli presented in the peripheral visual field (i.e., under conditions that engage the subcortical systems), but not when they are displayed in the central visual field.
The fact that newborns just hours to days after birth look longer at the mother’s face than at a stranger’s (even when cues from her smell and voice are eliminated) indicates that, from very early on, there is a mechanism capable of learning about individual faces based on experience. This early learning system may be mediated by early developing allocortical areas known to be involved in memory (see earlier discussion; Shaw et al., 2008). Nevertheless, the face-processing abilities of the newborn are qualitatively different than the sophisticated capacities of adults, and none of these tendencies are necessarily the direct precursors of the cortically mediated, adult face-processing system (see Chapter 44). Instead, these responses might serve the purpose of providing input to developing cortical circuits that will at some later time functionally emerge to mediate face processing.
At 3 Months of Age Infants Are Able to Form Categories of Visual Stimuli
A marked change in infants’ visual attention to faces occurs at approximately 8 weeks of age. At this time, infants’ preferential following of peripheral moving faces declines, and a preference emerges for fixating faces compared to other patterns presented in the central visual field. This behavioral change is thought to be due to the functional development of visual cortical pathways that inhibit the preferential following response and mediate the new preferential fixation response. At 2 months of age, infants become more sensitive to the internal facial features of static faces. In addition, within the face, the eyes are a more salient feature than the nose or mouth, but where the eyes are located is immaterial to the babies’ preference for face-like drawings. At roughly the same age, infants begin to relate information between individual faces and to form categories; that is, they perceive visual stimuli with comparable features as being more similar than visual stimuli with different features. They also exhibit inversion effects (i.e., recognition of upright faces is easier than recognition of inverted faces). The change between 1 and 3 months has been associated with the functional development of temporal cortical areas and their connections with the hippocampus and adjacent structures. However, at this early age, the change may not be specific to faces, as 3-month-old infants can form perceptual categories of a variety of other complex categories, such as tables and trees.
During childhood, the child’s prototype of the face becomes more tuned to the types of faces that the child sees most often, and with this may emerge race effects and other species effects seen in adult face processing. Other race effects refer to the ability to distinguish among faces of one’s own race more quickly than faces of other races. Other species effects refer to the ability to distinguish between human faces more quickly than faces of other species. These last changes may be based on experience due to increased exposure to faces, the number and type of features children attend to and encode, and the maturation of a posterior temporal cortical area (the fusiform area) that is known to be involved in face processing in adults.
Fear of Unfamiliar Faces Emerges between 7 and 9 Months
Although infants are able to discriminate among some facial expressions of emotion by 3 months of age, they do have difficulty discriminating between sadness and surprise. It is only by 7 months of age that infants, like adults, show categorical perception of facial expressions. For example, with pictures of faces in which emotional expressions are progressively degraded from happy to fear, human infants and adults exhibit more accurate discrimination for pictures of faces that cross emotional categories (happy versus fear) than for pictures of faces within the same emotional category, despite equal physical differences in the pictures. With this increased ability to categorize faces, and the increased exposure to familiar faces, two remarkable events occur between 7–9 months of age: the emergence of fear for unfamiliar people and anxiety during temporary separation from the caretaker. Distress under both conditions must involve more than the ability to discriminate strangers from parents because 3 month-olds can make this discrimination, but do not show fear reactions.
One speculation concerning stranger and separation fear is that improvements in working memory, and an enhanced ability to retrieve memories of a past event, are required for the appearance of these reactions. This change appears to be associated with the maturation of the orbital frontal cortex (Schore, 1996), a region of the prefrontal cortex intimately interconnected with limbic areas in the temporal lobe (temporal pole cortex and amygdala), with subcortical drive centers in the hypothalamus, and with dopamine neurons in reward centers in the ventral tegmental area. By the end of the first year, the initial phase of orbital frontal maturation is achieved and allows for developmental advances that enable the individual to react to situations on the basis of stored representations, rather than on information immediately present in the environment. In the case of the orbital frontal cortex, this capacity applies specifically to socioemotional information. Indeed, by 10 months, infants are first able to construct and store abstract prototypes of human visual facial patterns, and can use these prototypes to evaluate novel information.
In an illuminating experiment, a preferential looking task was used to habituate 10-month-old infants to pictures of faces. One condition presented faces of different females, and in the other condition, the same female face was presented in various poses. After infants habituated to the stimuli (i.e., decreased their looking), they were given two test trials: one with a face of a familiar female and another with a face of a novel female. When infants were previously habituated using different female faces, they spent equivalent amounts of time gazing at familiar and novel faces on the test trials. However, when infants were habituated to the same face with different poses, they generalized their looking response to the familiar face but dishabituated (or looked longer) at the novel face. This pattern of results suggests that infants can abstract relevant categorical information by 10 months of age.
Changes in Visual Search Are Related to Development of the Prefrontal Cortex
Infants younger than 7 or 8 months of age will not uncover a hidden object. If a cloth is thrown over a toy while an infant of 5 or 6 months is reaching for it, the infant will withdraw his or her hand and stop reaching. By 7 or 8 months, most infants who watch an object being hidden can retrieve it. However, if the infant then watches as the object is hidden at a second location, most infants of 7–8 months search for the object at its first hiding location, which is now empty. Jean Piaget called this the A-not-B error because the infant is correct at the first hiding place (A) but not at the second (B). It is rare to see this error when there is no delay between when the object is hidden and when the infant is allowed to reach. However, a delay as brief as 1, 2, or 3 s is sufficient to produce the error in infants 7–8 months of age. As they grow older, the error is still seen, but only if the delay between hiding and retrieval is increased. The A-not-B error suggests that during the second half of the first year the brain has matured sufficiently to enable the infant to hold a representation of the object’s location in mind (working memory) for a few seconds (see Chapter 50) or to understand the relationship of (a) their previous action of retrieving the object, (b) the subsequent hiding of the object in a different location (even though they observed the hiding), and (c) the object’s present whereabouts (Diamond, 1990). The enhanced working memory observed in the 7- to 10-month-old infant parallels the protracted maturation and refinement of dorsolateral prefrontal cortex circuitry (Fig. 43.3). After 12 months of age, it is difficult to elicit the A-not-B error.
Language Undergoes Rapid but Prolonged Development
In humans, the prolonged structural, metabolic, and neurophysiological maturation of “association” cortical areas, which continues well into adolescence, provides the substrate for the panoply of higher cognitive functions that continue to develop during this time. Attempts have been made to link the very rapid development of speech and language skills over the first 3 years of life to these general changes. This goal has remained elusive, probably because so many aspects of the brain and behavior are changing together. Moreover, key elements of language appear to be processed at cellular and synaptic levels of organization that are difficult to investigate in humans with even the most sophisticated techniques available.
There is wide agreement that language is strongly dependent on structures within the left perisylvian region (see Chapter 49). Very early on (by 28 weeks of gestation), structural asymmetries appear between the temporal lobes; these may provide the substrate for the functional asymmetries that appear later. The rapid and early acquisition of phonological information and speech production and comprehension, and the subsequent burst in size of the vocabulary, may be linked to the rapid rise in the number of synapses and the marked increases in cortical metabolism that occur during the second year of life. Also, the persistence of large numbers of exuberant synapses through adolescence may provide the anatomical substrate for prolonged neural plasticity and recovery of language skills following cortical damage in the first decade of life. It is well established that language skills can display considerable recovery following large lesions involving the left hemisphere during the first 7–10 years of life. The impressive language recovery in children who have undergone left hemisphere removal is even more striking in light of the enduring impairment seen in specifically language-impaired children or the reading difficulties encountered by dyslexics, in whom macroscopic aspects of brain structure are basically normal. These findings underscore the importance of characterizing microscopic structural aspects of the brain and the functional organization of the brain in relation to processing.
The prolonged time course of development that may confer plasticity on the immature brain appears to be characterized by optimal or critical periods (see Chapter 21) for language acquisition. Several studies report that both first and second language acquisitions are impaired, and cerebral organization is altered, when language is acquired after the first decade of life. Moreover, as has been observed for vision (Fig. 43.2), different aspects of language appear to display different critical periods. Vocabulary items can be acquired long past the first decade of life, but the grammatical rules of a language appear to be acquired most readily before the age of 10. Along with other evidence, this pattern suggests that different neural systems, with differing developmental time courses, mediate these various aspects of language.
Summary
Cognitive development parallels some of the most important changes in brain development. Thus, the visual abilities of the newborn are limited and control by subcortical systems predominates. With the maturation of visual cortical areas in the first few postnatal months, visual acuity, visual alertness, and stereopsis develop. With further development of visual cortical areas within the temporal lobe, an infant’s ability to recognize faces using internal facial features, mostly the eye region, emerges around 3 months of age. However, it is only by 7–9 months of age that infants develop fear of unfamiliar people. It is believed that this behavior is associated with the development, around this age, of the orbital frontal cortex, a cortical area that enables the infant to react to situations on the basis of stored representations (working memory). The enhanced spatial working memory abilities observed by the middle of the first year parallel the protracted maturation and refinement of the dorsolateral prefrontal circuitry. Finally, the maturation of association areas of the cortex, which continues well into adolescence, provides the substrate for the development of higher cognitive functions, such as language. In the future, it will be possible to analyze the neural basis of cognition in increasing detail by using high-resolution methods for imaging brain structure and function in normally developing children and children with specific structural or functional deficits.
Cognitive Aging
Average Human Life Expectancy has Increased Dramatically
The world’s population is growing older. Whereas life expectancy in the United States was approximately 50 years in 1900, infant mortality rates decreased dramatically during the last century, and combined with advances in disease prevention and treatment, current life expectancy is approaching 80 years in many industrialized countries. A significant demographic trend is that the percentage of the population over 65 years of age is projected to more than double between the years 2000 and 2030, with particularly rapid growth among those over 85. Neurodegenerative disorders such as Alzheimer’s disease are among the most devastating consequences of growing older (see section on Dementia, this chapter). There is increasing recognition, however, that many otherwise healthy aged individuals experience deficits in memory and other aspects of cognitive function that, although less severe, pose a significant risk to independence and the quality of life. The following sections outline key concepts from the study of normal cognitive aging in the absence of frank disease.
Effects of Normal Aging on Memory Are Variable
Cognition encompasses a variety of dissociable capacities, including perception, attention, executive function, and memory (Section VII, this volume), mediated by partially distinct neural systems. On the basis of this organization, neuropsychological research documenting the specific types and severity of cognitive deficits that accompany aging can provide important clues about the brain systems that mediate dysfunction. This strategy has been widely exploited to examine the effects of normal aging on learning and memory, where a rich background is available concerning the characteristics of impairment that follow damage to selective brain regions in young adults.
A key concept to emerge from neuropsychological studies of aging is that the status of memory mediated by the hippocampal formation and related medial temporal lobe structures varies substantially among older individuals (Fig. 43.4). In humans and animal models, memory ability is continuously distributed across a broad range such that some aged individuals exhibit considerable impairment while others—at the same chronological age—perform as well as young adults. Long considered a complicating factor in gerontological research, this variability suggests the important conclusion that marked deterioration in memory is not an inescapable consequence of growing older and that the functional integrity of the medial temporal lobe system is preserved in a substantial proportion of aged individuals. The next section considers findings from animal models supporting this perspective, and that have guided efforts to identify the neurobiological alterations reponsible for individual differences in normal cognitive aging.
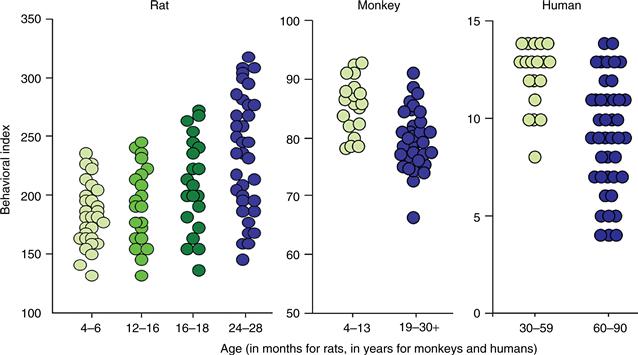
Figure 43.4 The status of memory varies considerably across aged individuals. The data comprise a summary measure of spatial memory in rats (courtesy of M. Gallagher, Johns Hopkins University), object recognition memory in monkeys (courtesy of P. Rapp, National Institue on Aging), and delayed recall in humans (courtesy of M. Albert, Johns Hopkins School of Medicine). For rats, low scores represent better memory, and high values reflect better performance in monkeys and humans. Symbols signify scores for individual subjects. Note that in all species the status of memory among the aged is distributed across a broad range, from individuals that score on a par with the best young adults to other individuals that exhibit substantial impairment.
Animal Models of Normal Neurocognitive Aging
As described in Chapter 48, normal adult rats solve the spatial, or “place,” version of the Morris water maze by learning and remembering the escape location in relation to cues surrounding the testing apparatus, and this capacity requires the hippocampal formation. Guided by this background, many laboratories have used the Morris water maze and other spatial tasks to examine the effects of aging on learning and memory mediated by the hippocampal system (reviewed in Rosenzweig and Barnes, 2003).
Aged rats learn the hidden platform location in the place version of the water maze more slowly than young adults. Although these findings are consistent with a possible hippocampal contribution to cognitive aging, an alternative account is that declining visual acuity, motor function, or other impairments give rise to performance deficits in older subjects. This possibility has been tested using control procedures that make many of the same sensory, motor, and motivational demands as the hidden platform task, but that lack an explicit spatial learning component. In a common task variant, for example, rats swim to a platform that protrudes above the surface of the water, providing a salient visual goal to guide navigation. Because the cued platform location is varied randomly across trials, learning and memory for spatial information are not required. Many aged rats acquire this task normally, demonstrating that they can swim proficiently, are motivated to escape, and retain sufficient visual acuity to support accurate cue-directed navigation. Thus, whereas some aspects of motor and visual function are indeed vulnerable to aging, these impairments fail to explain the deficits aged rats exhibit in the spatial version of the water maze. Instead, the overall profile observed in aging—impaired spatial learning and memory against a background of intact goal-approach learning—is qualitatively similar to the effects of damage to the hippocampal system in young subjects. Other tasks that require hippocampal integrity are also sensitive, confirming that learning and memory mediated by this system are susceptible to age-related decline (Rosenzweig & Barnes, 2003).
As noted earlier, research in animal models has revealed substantial individual variability in the cognitive outcome of aging. Figure 43.4, for example, illustrates individual learning scores for large numbers of young, middle-aged, and aged rats tested in the spatial version of the water maze. The performance measure in this case reflects distance from the escape platform, averaged over multiple trials, and accordingly, low scores represent better learning, with searching focused near the escape location. Similar to the distribution of memory scores for people (Fig. 43.4), spatial learning capacity among aged rats varies across a broad range such that some perform as well as even the most proficient young subjects. By comparison, approximately half of the older animals exhibit marked deficits, scoring outside the range of the young adult group.
Variability in the cognitive effects of aging has been examined in nonhuman primates as well, taking advantage of testing procedures adopted from studies on the effects of medial temporal lobe damage in young adult monkeys (see Chapter 48). In the delayed nonmatching to sample task, subjects are tested for their ability to recognize a visual stimulus after a delay ranging from seconds to many minutes or more. Monkeys with sufficiently extensive damage to any component of the medial temporal lobe memory system display significant deficits on this task, particularly under conditions of increased memory demand, with long retention intervals. Recognition memory declines during aging in the monkey, and as in humans and rats, the degree of impairment varies substantially across individual subjects (Fig. 43.4). As discussed in the next section, this variability makes it possible to ask what neurobiological alterations distinguish the brains of individuals with age-related memory impairment from other, age-matched subjects with preserved function. The aim by this approach is to identify changes in the structure and physiology of the aged brain that are specifically coupled to individual differences in the cognitive outcome of normal aging.
Neurobiology of Age-Related Memory Decline
Age-Related Learning and Memory Impairment Is Associated with Compromised Hippocampal Physiology
Behavioral studies suggest that the functional integrity of the hippocampal system declines during aging. Electrophysiological investigations extend these observations by directly assessing the computational processing functions of the aged hippocampus. Models of learning-related cellular plasticity, such as hippocampal long-term potentiation and depression (LTP and LTD; see Chapter 47), establish a useful background against which to explore the effects of aging. A prominent theme is that aging influences hippocampal physiology in a highly selective manner, sparing many aspects of function (reviewed in Burke and Barnes, 2006: and Wilson, Gallagher, Eichenbaum, and Tanila, 2006). Parameters that exhibit little or no change in older subjects include the resting potential, input resistance, and the amplitude and duration of evoked action potentials of principal hippocampal neurons. By comparison, although the peak magnitude of LTP is comparable in the young adult and aged hippocampus, the intensity and frequency of stimulation necessary to achieve that response increase with advanced age. Once established, LTP also decays to prepotentiated baseline levels more rapidly in aged subjects than in young animals. This enhanced rate of decay is correlated with the rapid forgetting aged rats exhibit on tests of spatial memory, consistent with the conclusion that a reduced capacity for synaptic enhancement in the hippocampus contributes to this behavioral deficit. Hippocampal LTD is also affected, demonstrating that multiple plasticity mechanisms are altered in relation to variability in the cognitive outcome of aging.
Electrophysiological studies of ongoing neuronal activity in awake-behaving rats provide a direct window on how normal aging influences hippocampal information processing. As described in Chapter 48, a particularly well-characterized phenomenon is that the firing rate of individual pyramidal neurons in the hippocampus increases dramatically as animals navigate through a restricted area within a testing environment. This “place field” activity is largely independent of the specific behaviors that accompany exploration and instead reflects the cognitive demands of testing in relation to sensory cues that define a particular spatial location. Recapitulating a theme from studies of LTP and LTD, many aspects of place field firing are unchanged in the aged hippocampus, including the percentage of neurons that exhibit location-specific firing, and the spatial selectivity of place fields. The overall scope of information encoded, however, is reliably altered (Burke & Barnes, 2006; Wilson et al., 2006). For example, hippocampal pyramidal cells in old rats are abnormally prone to confuse familiar environments that include overlapping stimulus elements, unpredictably engaging the wrong distribution, or “map,” of place fields during successive bouts of exploration in a given setting. Hippocampal encoding also becomes increasingly rigid with age such that a relatively narrow subset of available spatial cues comes to control place field activity. These changes are more prevalent in the CA3 field of the hippocampus than in CA1, and among aged individuals that demonstrate impaired spatial learning (Wilson et al., 2006). Research on the structural integrity of the hippocampus, discussed in the following sections, points to a potential link between the behavioral and electrophysiological consequences of aging.
Early Findings Suggested Neuron Loss Is Distributed Diffusely throughout the Aged Brain
One of the most widely held notions about brain aging is that substantial numbers of neurons inevitably die as we grow older. Early studies supported this view, suggesting that neuron death occurs throughout life, with the cumulative loss exceeding 50% in many neocortical areas by age 95 (for an historical review, see Brody, 1970). Although the magnitude of effect appeared to vary, significant age-related neuron loss was reported for all regions examined, including both primary sensory and association areas of the cortex. On the basis of these early observations it seemed reasonable to suppose that widely distributed neuron death might account for many of the cognitive deficits associated with normal aging. A significant concern, however, was that the finding of widespread neuronal loss might be attributable to preclinical dementing disease in an unknown proportion of the aged individuals who were examined. This confound is overcome in studies using animal models of cognitive aging because rats and monkeys do not develop dementia spontaneously. In addition, as described next, improved quantitative tools for examining neuron loss in the aged brain are now widely available.
Memory Impairment during Normal Aging Does Not Require Substantial Hippocampal Neuron Loss
Methodological developments in quantifying cell number have prompted significant revision in traditional views on age-related neuron loss (Morrison & Hof, 1997). Before the 1990s, most investigators focused on cell density, defined as the number of neurons present in a fixed area or volume of tissue. A significant limitation, however, is that density can vary widely in the absence of any actual difference in cell number. Assume, for example, that total neuron number is identical in two brains, but that the overall size of the brains differs due to normal biological variability among individuals, gliosis, white matter abnormalities, or other neuropil alterations. Under these conditions, neuron density will be lower in the larger brain, simply as a consequence of the cells being distributed in a larger volume. In this way, real or processing-related volumetric differences between young and aged brains could substantially influence cell density in the absence of any actual age-dependent difference in neuron number.
The field of stereology has provided standardized tools for quantifying total neuron number in any defined brain region, yielding an unequivocal measure for examining potential neuron loss during normal aging (see Box 43.1). In contrast to early studies measuring cell density, the conclusion from investigations using modern stereological methods is that the total number of principal neurons (i.e., granule cells of the dentate gyrus and pyramidal neurons in the CA3 and CA1 fields) is preserved in the aged hippocampus. Substantial sparing is also seen in adjacent parahippocampal cortical regions that critically participate in memory. Whereas similar results have been observed in humans, data from animal models are particularly illuminating in demonstrating that hippocampal neuron number remains stable even among aged individuals with pronounced learning and memory deficits. As illustrated in Figure 43.5, for example, stereological analysis reveals that the total number of neurons comprising the hippocampal region is comparable in young adult subjects and aged rats, regardless of their capacity for spatial learning and memory.
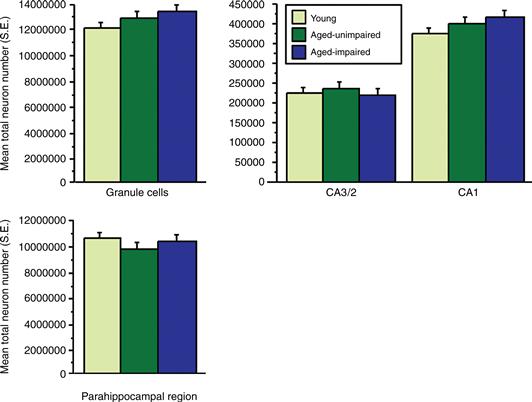
Figure 43.5 Estimated total neuron number (plus S.E.) in principal cell layers of the hippocampus and parahippocampal cortices (i.e., entorhinal, perirhinal, and postrhinal) for behaviorally characterized young and aged rats. All values are unilateral. Half of the aged rats exhibited substantial spatial learning deficits in the Morris water maze (aged impaired); the other half performed within the range of learning scores for the young group (aged unimpaired). Neuron number is stable with age and across a broad range of learning capacities. Adapted with permission from Rapp and Gallagher (1996) and Rapp, Deroche, Mao, and Burwell (2002).
The dentate gyrus of the hippocampus is one of only a few niches of neural stem cells capable of generating new neurons in the adult brain (see Box 14.1). Whereas diffuse neuron death is no longer considered a prominent feature of normal aging, considerable interest centers on the idea that a decline in hippocampal neurogenesis contributes to cognitive aging, and that interventions designed to enhance neurogenesis might protect against age-related memory decline, or even rescue function in Alzheimer’s disease. Although neurogenesis rates decrease precipitously in the adult rat hippocampus, this effect emerges much earlier in the lifespan than age-related deficits in hippocampal memory, and it fails to predict individual variability in the severity of impairment (reviewed in Bizon and Gallagher, 2005). Considerable additional study will be needed in order to fully exploit neural stem cell biology in support of healthy cognitive aging.
Hippocampal Connectivity Is Compromised during Normal Aging
Substantial research has focused on changes in synaptic connectivity and other markers of functional deterioration as potential contributors to normal cognitive aging. The entorhinal cortex originates the major source of cortical input to the hippocampus, giving rise to the so-called perforant path, which synapses on the distal dendrites of dentate gyrus granule cells in outer portions of the molecular layer. The same entorhinal cortex neurons also innervate other fields of the hippocampus, including the most distal aspects of CA3 pyramidal cell dendrites. Multiple lines of investigation suggest that the integrity of these inputs is compromised with age. Stereological quantification of electron microscope images, for example, has documented that aging is associated with significant loss among a morphologically distinct subset of synapses in the outer molecular layer of the dentate gyrus (i.e., the zone innervated by the entorhinal cortex) (Geinisman, de Toledo-Morrell, Morrell, Persina, & Rossi, 1992), together with alterations of synaptic structure (e.g., descreased postsynaptic density length) in other hippocampal subfields. Input originating in the entorhinal cortex conveys much of the neocortically derived information that the hippocampus processes in support of normal memory, and accordingly, it is reasonable to suppose that the disruption of entorhinal–hippocampus connectivity might contribute to the cognitive outcome of aging. Consistent with this proposal, the magnitude of morphological alterations observed in the hippocampal termination zones of the entorhinal cortex is greatest among aged animals with documented deficits on tasks that require the hippocampus and among older rats that exhibit abnormalities in various physiological measures of hippocampal plasticity.
Box 43.1 The Optical Fractionator Stereological Method
The figure shows the key features of a stereological technique designed to provide accurate and efficient estimates of total neuron number in a brain region of interest. The hippocampal formation is used as an example. The method consists of counting the number of neurons in a known and representative fraction of a neuroanatomically defined structure in such a way that each cell has an equal probability of being counted. The sum of the neurons counted, multiplied by the reciprocal of the fraction of the structure that was sampled, provides an estimate of total neuron number.
Serial histological sections are prepared through the rostrocaudal extent of the hippocampus and are stained by routine methods for visualizing neurons microscopically. An evenly spaced series of the sections is then chosen for analysis (positions represented schematically in top panel). This first level of sampling, the “section fraction,” therefore comprises the fraction of the total number of sections examined. For example, if every tenth section through the hippocampus is analyzed, the section fraction equals 1/10. The appropriate sections are then surveyed according to a systematic sampling scheme, typically carried out using a microscope with a motorized, computer-controlled stage. The lower part of the figure illustrates this design in which the microscope stage is moved in even X and Y intervals, and neurons are counted within the areas defined by the small red squares (“a (frame)” in the inset). The second level of the fractionator sampling scheme is therefore the “area fraction,” or the fraction of the XY step from which the cell counts are derived.
The last level of sampling is counting cells only within a known fraction (h) of the total section thickness (t), avoiding a variety of known errors introduced by including the cut surfaces of the histological preparations in the analysis. This is accomplished using a high-magnification microscope objective (usually 100X) with a shallow focal depth. In the illustration provided, the “thickness fraction” is defined as h/t. Neurons are counted as they first come into focus, according to an unbiased counting rule, called the “optical disector,” that eliminates the possibility of counting a given cell more than once.
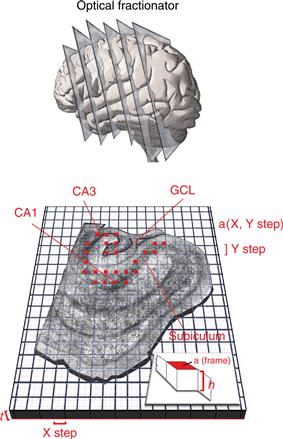
Finally, total neuron number in the region of interest (N) is estimated as the sum of the neurons counted (sumQ−), multiplied by the reciprocal of the three sampling fractions; the “section fraction,” “area fraction,” and the “thickness fraction.” For the present example, the total estimated neuron number is given by
Original illustration design by L. E. Mekiou and P. R. Rapp.
Peter R. Rapp
Executive Function Mediated by the Prefrontal Cortex Declines during Normal Aging
Normal aging is sometimes characterized as a generalized process of nonspecific deterioration across many neural systems, implying that age-related cognitive decline might be the cumulative effect of subtle alterations distributed diffusely throughout the brain. Little empirical evidence actually supports this notion, however, and as we have seen in the case of hippocampal circuitry, the current perspective is that the neurobiological consequences of aging exhibit considerable selectivity. At the same time, it is important to recognize that the effects of aging on memory do not occur in isolation and that cognitive capacities mediated by other neural systems are also vulnerable.
As discussed in Chapter 50, the functionally and neuroanatomically heterogeneous areas comprising the prefrontal cortex support a constellation of capacities referred to as “executive function.” Whereas damage to these regions does not produce the frank amnesia that follows medial temporal lobe lesions, significant deficits are observed in a variety of processing functions that influence memory, including the strategic use and manipulation of remembered information, the ability to recall the source from which information was acquired, and the order in which it was presented. It is therefore of interest that a qualitatively similar profile of deficits emerges during the course of normal aging (reviewed in Fletcher and Rapp, 2012). Older people, for example, often experience difficulty recalling the original source of remembered information, even when recollection of the target item itself is successful. These failures in “source memory” predict performance on other tests of frontal lobe function, including memory for temporal order, suggesting they share a common neurobiological basis.
Neuropsychological studies in animal models reinforce the conclusion that prefrontal cortex dysfunction contributes to certain features of cognitive aging. One of the best-documented deficits associated with aging in monkeys, for example, is poor performance under testing conditions that emphasize the spatial and temporal, or the “where and when,” components of memory (Fletcher & Rapp, 2012). In the classic delayed response task, subjects observe while a food treat is hidden in one of two locations that are then covered with identical plaques (see Fig. 50.5). Because each reward location is baited frequently across trials within a single test session, there is substantial opportunity for interference and accurate performance depends on the ability to keep in mind the spatial location that was baited most recently. Damage involving the dorsolateral prefrontal cortex in young monkeys produces impairment on the delayed response procedure at retention intervals of just a few seconds or more. Deficits on this task following lesions of the hippocampal system, in contrast, are selectively observed at longer delays. Numerous studies since the late 1970s have documented that aged monkeys exhibit a profile of delayed response impairment qualitatively similar to the effects of prefrontal cortex lesions in young animals. Additional signs of prefrontal decline are also found in aged monkeys, including marked perseveration and behavioral rigidity when subjects are challenged with shifting task contingencies. For example, whereas young adults quickly cease responding to a previously positive stimulus when it is no longer rewarded, aged monkeys are slower to make this behavioral adjustment.
Are the effects of normal aging on prefrontal cortex capacities coupled to other signatures of cognitive decline, consistent with the view of aging as a process of generalized deterioration? Available evidence suggests that this is not the case. For example, age-related deficits on tests of prefrontal cortex function tend to emerge earlier during the course of aging than alterations in declarative memory supported by the medial temporal lobe system. In addition, when impairments indicative of both frontal and medial temporal lobe dysfunction cooccur in the same subjects, these effects are statistically unrelated to each other (reviewed in Fletcher and Rapp, 2012). Thus, different aspects of cognitive function can decline independently during aging and these effects are not the consequence of a global, brain-wide degenerative process.
Neurobiology of Aging in the Prefrontal Cortex
The basis of age-related decline in capacities supported by the prefrontal cortex is the subject of intense investigation. Similar to the hippocampal system, neuron death is not a prominent feature of normal aging in many frontal lobe areas that mediate information processing functions known to decline with age. Other structural alterations have been documented, however, including a substantial decline in the density of prefrontal cortex synapses, marked changes in dendritic architecture, and a variety of abnormalities in the myelination of axons (Peters, Morrison, Rosene, & Hyman, 1998). These parameters are correlated with behavioral signatures of cognitive aging in monkeys, raising the possibility that, like the hippocampal system, changes in the functional organization of critical prefrontal networks may play a significant role. Consistent with this possibility, recent findings document that delayed response impairment in the aged monkey is associated with the blunting of memory-related neuronal activity in the dorsolateral prefrontal cortex, and that normal task dependent firing is at least partly restored by pharmacological treatments that benefit behavioral performance (Wang et al., 2011).
Results from in vivo imaging studies in humans complement the animal model data and further suggest that the functional organization of the aged brain is more dynamic than previously presumed. The traditional understanding has been that successful cognitive aging reflects the persistence of a youthful condition, dependent on the same mechanisms that support normal function in younger individuals. Rather than following a simple pattern of regional decline or maintenance, however, growing evidence indicates that aging is accompanied by reliable change in the distribution of activations across brain areas important for normal cognitive function, including the prefrontal cortex and medial temporal lobe. Indeed shifts in the balance of regional activation are observed even when memory is tested under conditions where aged individuals score as accurately as young adults. The implication of these findings is that the overall network engaged during testing is significantly altered and that successful cognitive outcomes in normal aging may rely, in part, on adaptive functional reorganization (reviewed in Hedden and Gabrieli, 2005). Intervention aimed at harnessing this dynamic capacity in support of optimally healthy cognitive aging is an area of active investigation.
Neurochemically Specific Subcortical Systems Are Susceptible to Aging and Positioned to Broadly Influence Behavior
Current evidence challenges the view that normal aging is associated with generalized deterioration distributed diffusely throughout the brain. Aging does, however, affect the integrity of neurochemically specific classes of cells whose ascending projections influence widespread brain areas. Cholinergic cell groups in the basal forebrain have been studied particularly intensively because this system exhibits pronounced degeneration in Alzheimer’s disease. Less severe effects are also observed during normal aging, involving acetylcholine-containing neurons that project to the hippocampus, amygdala, and neocortex. Cholinergic cell loss might therefore disrupt the information processing functions of these target regions, and consistent with this possibility, reliable correlations have been reported between the magnitude of cholinergic deficits and behavioral impairment in aged individuals. Cholinergic abnormalities alone, however, fail to account for the full profile of cognitive deficits observed during aging since neurotoxin lesions that selectively destroy these neurons fail to reproduce key features of the behavioral decline observed in older individuals. One possibility is that age-related cholinergic impairment comprises an important component of a broader constellation of alterations that leads to cognitive dysfunction. Other neurochemical systems that might contribute include dopamine-containing circuitry that originates in midbrain cell groups and noradrenergic inputs that arise from the locus coeruleus. These systems innervate a variety of more anterior brain regions, and consistent with this broad distribution, neurochemical alterations have been linked to a diverse array of behavioral deficits ranging from age-related cognitive impairment to disturbances in motor function. These findings encourage the view that appropriately designed pharmacological interventions might have beneficial effects on a similarly broad profile of outcome measures of aging.
Summary
This section considered the cognitive and neurobiological effects of normal aging in humans and animal models. Key themes include the observation that the impact of aging on memory varies widely from one individual to the next. Whereas neuron death was once presumed to be the proximal cause of many cognitive deficits associated with aging, including learning and memory impairment, it is now clear that such decline does not require marked neuron loss in the hippocampus and related cortical areas. Instead, subtle alterations in connectivity, and downstream changes in cellular function, are more likely causative factors. Fueled by advanced in vivo brain imaging studies, a comprehensive, neural systems account of normal cognitive aging is beginning to emerge.
Pathological Processes in Cognitive Development and Aging
Developmental Psychopathologies
Research in this field focuses on elucidating the interplay among the biological, psychological, and social-contextual aspects of normal and abnormal development across the life span. Recent advances in neuroimaging and gene linkage technology have vastly expanded research possibilities for noninvasive clinical studies. Even with a new arsenal of research technologies, however, it has not been possible to isolate and define characteristics of each developmental disability. Such efforts are limited by several factors:
1. Disagreement regarding the criteria for diagnosis. Because different researchers often use differing behavioral criteria, cross-study comparisons frequently show inconsistencies in results.
2. Heterogeneity of disorders. Even when consistent behavioral criteria are carefully applied, different underlying etiologies can result in the same behavioral profile.
3. Difficulties inherent in the study of children. Although modern brain imaging techniques such as magnetic resonance imaging are considered noninvasive, they are stressful and time-consuming, and parents will not always allow their affected children to participate in such studies. Although studies can be conducted using adults who have been affected since childhood, data obtained from such studies may not accurately reflect anomalies that characterize the early disruption of brain development because brains change over time. Moreover, retrospective diagnoses of childhood disorders frequently rely on memory (e.g., a patient is asked, “Did you have difficulty reading as a young child?”) and thus can be unreliable.
Despite these challenges, ongoing research has uncovered important neurological and genetic features that seem to be associated with specific developmental disorders (see Box 43.2). The following sections outline what is known about how specific injuries or anomalies in development of the brain may result in specific patterns of cognitive and behavioral impairment. Two examples of developmental psychopathology are described: autism and schizophrenia.
Box 43.2 Dyslexia
Reading skills occupy a uniquely important position in overall cognitive development, strongly dependent on antecedent skill acquisition and critically supportive of educational success. After accounting for the effects of inadequate instruction, the most common cause of severe reading problems in childhood is developmental dyslexia, a disorder typically characterized by impairment in applying the sound–correspondence rules necessary to decode print (see Chapter 49). Although reading impairment is common, with an incidence of 5 to 10% among schoolage children, our understanding of its biological roots is relatively recent.
Neurobiological studies of dyslexia are challenging, as the behavioral manifestations of this disorder are complex. Although defined by poor reading performance, dyslexia involves additional deficits, including poor phonological processing, poor verbal working memory, and slow naming ability. Dyslexics frequently show subtle deficits in motor control and early sensory processing, most notably in the visual system. Imaging studies have revealed that dyslexic brain structure differs from that seen in individuals with normal reading skills. Micro and macroscopic structural abnormalities have been detected in perisylvian language regions (temporal and parietal banks of the sylvian fissure as well as the insula), visual system structures, the thalamus, and the corpus callosum. The planum temporale is an expanse of neocortex on the temporal bank of the sylvian fissure, its anterior border defined by Heschl’s gyrus. From the time of birth the left planum temporale is larger than the homologous region in the right hemisphere in 70 to 80% of individuals. Reduction or reversal of this leftward asymmetry has been documented in individuals with developmental dyslexia. The functional concomitants of these anatomical variations have been explored in functional imaging studies that have revealed differences in patterns of task-related activity between dyslexics and controls during the performance of reading and phonological decoding tasks. Interestingly, the cortical areas identified are similar in English, French, and Italian dyslexics, supporting a biological origin for this reading abnormality, independent of cultural upbringing. Similarly, candidate genes for dyslexia have been identified and replicated in different countries, suggesting the multigenetic contribution to reading disability.
Besides the problems related to reading, dyslexics also exhibit subtle abnormalities in visual processing. Evidence for selective involvement of magnocellular pathways of the visual system (see Chapter 26) has accumulated, beginning with the demonstration that cell bodies in the magnocellular layers of the lateral geniculate nucleus are smaller in dyslexics compared to a control group. The fact that both contrast sensitivity and visual persistence are abnormal in reading disabled children is consistent with the notion that these children have disturbances in the magnocellular system, which is specialized for temporally demanding visual processing. These behavioral results are supported by functional imaging studies demonstrating less task-related activity in extrastriate and parietal cortex during visual motion detection by dyslexics.
Future studies will determine the nature of the neural mechanisms that link deficits in early visual processing with more cognitive skills such as phonological awareness. Also remaining to be clarified are issues concerning which of these deficits is a primary cause of dyslexia as opposed to a consequence of having partially compensated for a reading disorder. Such information will aid in identifying better avenues for reading remediation. To date, the best methods for improving the reading abilities of dyslexics focus on structured teaching of the code that allows individuals to sound out words (phonological awareness) in the context of fluency and comprehension exercises.
Guinevere F. Eden
Autism
Autism is a neurodevelopmental syndrome first described by Kanner in 1943, characterized by deficits in multiple domains including social interaction, play, language, and communication, and a restricted range of interests and activities, including repetitive and stereotyopic movements and persistent preoccupation that begin in infancy and become more apparent by the end of the third year of life. Although autism was once considered a rare disorder, recent surveys report a prevalence of 40 to 50 per 10,000 births. Parents often first become concerned because their child fails to use words to communicate, even though he/she recites passages from videotapes or says the alphabet. Like other disorders of abnormal cognitive development, autism is a heterogeneous condition that can range widely in severity.
Characteristics of Autism
A multitude of symptoms occur in autism, including social isolation (the autistic child largely ignores other people, shows little attachment to parents or other relatives, and retreats into a world of his or her own); stereotyped behaviors (the autistic child rocks back and forth, stares at neutral stimuli, rotates an object, or engages in other repetitive behaviors for long, uninterrupted periods); resistance to change in routine; abnormal responses to sensory stimuli (the autistic child may ignore visual stimuli and sounds, especially speech sounds, sometimes to such an extent that it might be thought the child is deaf); inappropriate emotional expressions (the autistic child has sudden bouts of fear and crying for no obvious reason; at other times the child displays utter fearlessness and unprovoked laughter); and poor use of speech. Individuals with autism can also exhibit symptoms that fall along a continuum of disorders. For example, many have mental retardation and show delays in the development of language. Others display neither language delay nor mental retardation, although they are clearly socially inept. Finally, autism is also associated with idiosyncratic interests and a restricted and repetitive behavioral repertoire. A growing number of studies over the last two decades have provided evidence that autism results from early onset brain dysfunction.
Neuropathological Alterations in Autism
Several studies of autistic individuals provide evidence for megalencephaly (whole brain enlargement) and for increased head circumference. The nature of this enlargement has yet to be studied carefully, but it is specifically observed in the occipital, parietal, and temporal lobes. Postmortem analyses of the brains of autistic individuals have revealed increased cortical thickness and abnormalities in cortical morphology. Furthermore, abnormalities in the size and density of neurons in the medial temporal lobe and limbic system, including the amygdala, hippocampus, and entorhinal cortex, septal nuclei, and cingulate cortex, have also been documented. These neuropathological abnormalities make sense with respect to certain major symptoms of autism because the limbic system is known to play a central role in coordinating social–emotional functioning. An fMRI study of autism has documented dysfunction localized to the ventral temporal lobe using a face recognition task (Baron-Cohen et al., 1999). In this study, high-functioning individuals with autism and matched controls were presented with a series of photographs of eyes while their brains were being scanned to assess brain regions activated by two different tasks. In the first task (experimental condition), each photograph was presented simultaneously with words (either unconcerned or concerned, or sympathetic or unsympathetic). Participants were asked to indicate by pressing a button which word best describes what the person in the photograph is feeling or thinking. In the second task (control condition), each photograph was presented with the words “male” or “female” and the subjects pressed a button to indicate gender. In both tasks, the performance of autistic individuals and controls was above chance. However, the controls were more accurate in the two tasks than the autistic individuals. To investigate the brain regions activated by the experimental task, brain maps obtained during the control task were subtracted from brain maps obtained during the experimental task. In the control group, the experimental task activated two main regions of the brain: the frontotemporal neocortical areas, including the left dorsolateral prefrontal cortex and supplementary motor areas, bilateral temporoparietal regions, and subcortical structures, including the left amygdala and parahippocampal gyrus. The autism group activated the frontal components but did not activate the amygdala. The significance of these data is that one critical role of the amygdala is to identify the mental states or emotions of other individuals and that this structure appears to be dysfunctional in persons with autism (see Pelphrey and Carter (2008), for additional information on the neuroanatomical substrates of social cognition dysfunction in autism).
Significant atrophy of the vermal lobules in the cerebellum has also been reported in autistic individuals, although this effect does not appear specific to autism and is found widely in neurodevelopmental disorders. By comparison, there is now extensive evidence for the involvement of higher association areas in the pathophysiology of autism, particularly the frontal and parietal cortices, as well as significant remodeling of cortical interconnectivity (Raznahan, Toro et al., 2010). Cytoarchitectural abnormalities that have been described in the cortex of patients with autism include a thickened cortex, high neuronal density, minicolumn alterations, the presence of neurons in the molecular layer, irregular laminar patterns, and poor gray-white matter boundaries.
Autism Is a Complex Genetic Disorder
The most compelling evidence for a genetic contribution to autism comes from studies of monozygotic (identical) and dizygotic (fraternal) twins. When one member of a pair of monozygotic twins is autistic, the other twin has more than a 50% probability of being autistic. However, if one member of a pair of dizygotic twins is autistic, the other twin has only a 3% probability of developing autism. Using conventional nomenclature, we say that monozygotic twins have a 50% or higher concordance for autism and that dizygotic twins have a 3% concordance. The relative risk factor for siblings is one of the highest for a complex genetic disorder. The most significant genetic finding relevant to autism is the recent identification of genes responsible for Rett’s syndrome and fragile X syndrome, two conditions associated with autistic features. Rett’s syndrome is caused by mutations in the methyl-CpG binding protein 2 (MECP2) gene. Decreased expression of this gene leads to a failure to suppress expression of genes regulated by methylation. Fragile X syndrome is a disorder caused by mutation of the gene FMR1 (see Box 43.3). A better understanding of how reduced expression of the MECP2 and FMR1 genes leads to mental retardation as well as social and language disorders will likely provide a better understanding of the autistic disorder. Over the last several decades, a growing list of rare mutations, such as NGLN4X, SHANK 3, NRXN1, have been described in individuals with autism (State, 2010). However, few of these genes are specific to autism and they instead variably contribute to genetic risk for a variety of neuropsychiatric disorders, including schizophrenia and attention-deficit hyperactivity disorder (ADHD). Although the findings provide tremendous promise, they also highlight the many challenges facing the study of the genetics of autism.
Box 43.3 Triplet Repeat Disorders
Not all neurogenetic diseases follow simple inheritance patterns; not all developmental disorders reveal themselves immediately; and diseases with similar genetic etiologies can have widely different manifestations. Trinucleotide repeat mutations, or dynamic mutations, were discovered only in the early 1990s but are responsible for a number of important neurodegenerative disorders, such as Huntington Disease (see Box 31.4), Fragile X mental retardation, and myotonic dystrophy. To date, 14 neurological disorders have been found to result from such a mutational mechanism, and the list will probably continue to grow.
Triplet repeat mutations are unstable and prone to expand as alleles are passed from one generation to the next, although contractions can occur. Repeats at different loci differ markedly in their rates and extents of expansion, and the instability of repeats at each locus often depends upon the sex of the transmitting parent.
Fragile X syndrome, an X-linked dominant mental retardation syndrome, was the first disease shown to result from a triplet repeat mutation. There is a CGG repeat expansion at a locus (Xq27.3) giving rise to a folate-sensitive fragile site (FRAXA). The CGG repeat ranges in length in the general population from 5 to 50 triplets. Affected individuals carry more than 230 repeats, up to several thousand triplets. CGG tracts between 45 and 200 repeats in length are termed “premutations”; they are not long enough to result in mental retardation, yet are prerequisite for further expansion to the full mutation. Escalating risk of affected offspring is associated with increasing size of premutations in female carriers. Curiously, offspring of male carriers of premutations are not at risk for Fragile X syndrome, as these alleles have not been observed to expand to full mutations upon male transmission. The normal function of the affected protein FMR1 remains to be fully determined, but it appears to play a role in RNA metabolism, likely in the areas of transport, stability, and/or translation.
Myotonic dystrophy (DM) remains a particularly enigmatic triplet repeat disorder affecting multiple systems, causing myotonia, muscle weakness, cardiac conduction defects, diabetes, cataracts, premature balding, and sometimes dysmorphic features and mild mental retardation. There is a tendency for these features to appear in more severe form in subsequent generations of a family. In DM there is a CTG repeat in the 3’ untranslated region of the myotonin protein kinase gene (DMPK). This CTG tract is small in the general population (5–37 triplets) but can expand dramatically to thousands of repeats. Slightly increased lengths (70–100) can result in mild effects, such as late-onset cataracts, and the classic symptoms of early adulthood myotonia are found in individuals with hundreds of repeats. Very large expansions can cause severe disease in newborns.
The mechanism(s) by which the mutation leads to the various clinical symptoms remains uncertain. A variety of effects have been described and the repeat expansions clearly affect both the DMPK gene and nearby genes. It is also likely that the expanded CTG repeat carrying mRNAs can act to diminish the levels of nuclear proteins that bind at these sequences, resulting in misregulation of genes not linked to DMPK. Mutations in equivalent mouse genes have not provided a complete model of the human disease. The phenotype in DM results from multiple effects of the repeat expansion, rather than a simple loss or gain of function mechanism.
Friedreich’s ataxia (FA) causes both limb and gait ataxia, loss of position and vibration sense, decreased tendon reflexes, cardiomyopathy, diabetes, and, in some patients, optic atrophy and deafness. FA is a recessive disorder and the only triplet repeat disorder known to involve a GAA sequence. Carriers of FA are rather common (1/85), and, consequently, so is the GAA expansion. Members of the general population carry between 7 and 34 repeats, while pathogenic alleles can number in the hundreds. The affected frataxin gene product appears to be involved in mitochondrial iron homeostasis, and its absence affects postmitotic cells rich in mitochondria. The mechanism by which the GAA repeat affects gene function appears to be inhibition of primary RNA transcripts.
A second class of triplet repeat disorders includes those caused by expansion of translated CAG repeats which encode a polyglutamine tract in each of the respective proteins. These “polyglutamine” disorders share many features, suggesting that a common pathogenetic mechanism is at play in spite of the fact that the mutated genes share no homology outside of the CAG repeats. They are progressive neurologic diseases with onset of symptoms in young to midadulthood with only a specific group of neurons vulnerable in each disease. Symptoms develop when the number of uninterrupted repeats exceeds approximately 35 glutamines. The phenotype is caused not by loss of function of the relevant protein but rather by a gain of function conferred by the expanded polyglutamine tract.
The spinocerebellar ataxias (SCA) are genetically distinct but overlap a great deal in their clinical presentation—between subtypes and even within families affected by the same disease. It is nearly impossible to distinguish one subtype from another on the basis of clinical signs alone. Spinocerebellar Ataxia Type 1 (SCA1) was the first of the inherited ataxias to be mapped. Patients with SCA1 suffer from progressive ataxia (loss of balance and coordination), dysarthria, and eventual respiratory failure. SCA1 is characterized pathologically by cerebellar atrophy with severe loss of Purkinje cells and brain stem neurons. Patients with SCA1 have an expanded “perfect” CAG repeat tract encoding ataxin-1, the function of which is largely unknown.
Studies of human patients with deletions of, for example, the HD, or SCA1 genes (and of mice lacking these genes) confirm that disease is not due to loss of function of the respective proteins. More importantly, studies of mouse and fruit fly models that over-express full-length or truncated forms of either ataxin-1, ataxin-3, huntingtin, AR, atrophin-1, or ataxin-7 demonstrate progressive neuronal dysfunction consistent with a gain of function mechanism. Several illuminating findings have emerged from the study of human patients, cell culture systems, and the various animal models. In transgenic mice, expression of truncated polypeptides containing expanded glutamine tracts caused widespread dysfunction that extends beyond the specific groups of neurons affected by the full-length protein. The expanded polyglutamine tracts may cause the respective proteins to misfold or resist degradation. Aberrant protein-protein interactions or alterations in levels of proteins essential for the normal functioning of neurons (such as proteins that regulate Ca+2 levels and neurotransmitters) could, in turn, exacerbate neuronal dysfunction and lead to neuronal death.
The finding that chaperone overexpression mitigates the phenotype in one vertebrate and several invertebrate models of polyglutamine disorders, and the identification of several additional modifiers of the neuronal phenotype, provide a platform for identifying key pathways of pathogenesis and potential therapeutic targets.
Adapted by Graham V. Lees with permission from Nelson, D., & Zoghbi, H. (2003). Disorders of Trinucleotide repeat expansions. In Encyclopedia of the human genome. London: Nature Macmillan.
Suggested Readings
1. Cummings CJ, Zoghbi HY. Trinucleotide repeats: Mechanisms and pathophysiology. Annual Review of Genomics and Human Genetics. 2000;1:281–328.
2. Wells RD, Warren ST, eds. Genetic instabilities and hereditary neurological diseases. San Diego: Academic Press; 1998.
Schizophrenia
Schizophrenia is a catastrophic illness with an onset typically in adolescence or early adulthood. It was identified and defined by Eugen Bleuler in 1911. The combination of significant incapacity, onset early in life, and chronicity of illness makes schizophrenia a particularly tragic disorder, occurring in 0.5 to 1% of the population.
Characteristics of Schizophrenia
The clinical symptoms of schizophrenia are often divided into two broad categories: positive and negative. Positive symptoms include delusions, hallucinations, disorganized speech, and disorganized or bizarre behavior. These symptoms are referred to as positive because they represent distortions or exaggerations of normal cognitive or emotional functions. The negative symptoms of schizophrenia reflect a loss or diminution of normal function. Negative symptoms include alogia (marked poverty of speech or speech devoid of coherent content), affective flattening (diminution in the ability to express emotion), anhedonia (inability to experience pleasure); avolition (inability to initiate or persist in goal-directed behavior), and attentional impairment. Although the positive symptoms of schizophrenia are often colorful and draw attention to the patient’s illness, the negative symptoms are an important feature of the disease that impair the person’s ability to function normally in daily life.
The profound and pervasive cognitive and emotional disturbances that characterize schizophrenia suggest that it is a serious brain disease affecting multiple functions and systems. Auditory hallucinations and disruptions in linguistic expression suggest involvement of the auditory cortex and perisylvian language regions. Other positive symptoms, such as delusions or disorganized behavior, are more difficult to localize to specific regions and suggest dysfunction distributed across multiple neural systems and circuits. Negative symptoms may be related to the prefrontal cortex, which mediates goal-directed behavior and fluency of thought and speech. The affective flattening and asociality suggest involvement of the limbic areas within the temporal lobe.
The Dopamine Hypothesis of Schizophrenia
A great deal of research since the 1950s has made it clear that schizophrenia is associated with abnormalities in the dopaminergic synapses of the brain. Still, the exact nature of this perturbation remains elusive. The strongest link between dopamine synapses and schizophrenia comes from studies of drugs that alleviate the symptoms of schizophrenia. A large number of antischizophrenic, or neuroleptic, drugs have been found, most of which belong to two chemical families, the phenothiazines, which include chlorpromazine, and the butyropherones, which include haloperidol. All of these drugs have two properties in common: they block postsynaptic dopamine receptors and they inhibit the release of dopamine from the presynaptic neuron. One interpretation of these results is that schizophrenia is due to excess activity at dopamine synapses. Establishing cause-and-effect relationships in human psychiatric disorders, however, is challenging. For example, although a substantial increase in the number of dopaminergic receptors is observed in the schizophrenic brain, because virtually all patients receive neuroleptic medication, it is not known if increased receptor density is a consequence of the disease itself or a response to the extended use of antidopaminergic drugs. An additional complication for the dopamine receptor hypothesis of schizophrenia concerns the time course for the effects of neuroleptic drugs. Although these treatments block dopamine receptors almost at once and reach their full pharmacological effectiveness within a few days, effects on behavior emerge more slowly, over 2 or 3 weeks. Clearly the symptomatic relief achieved with neuroleptic intervention involves mechanisms in addition to the blockade of dopamine receptors.
One possibility is that the prolonged use of neuroleptic drugs decreases the number of spontaneously active dopamine neurons in what is known as the mesolimbic system, a set of neurons that projects from the midbrain tegmentum to the limbic system. Another possibility is that the underlying problem in schizophrenia is not an excess of dopamine activity at all, but a deficit of glutamate activity. Glutamate is the predominant excitatory amino acid released by neurons in the cerebral cortex that project widely throughout the limbic system, and dopamine synapses are known to inhibit glutamate release in these target regions. If glutamate release is reduced in the schizophrenic brain, one way to correct this deficiency would be to block activity at dopaminergic receptors, relieving glutamate synapses from inhibition.
Neuropathological Alterations in Schizophrenia
The most consistently described structural alterations in schizophrenia are ventricular enlargement and decreases in the volume of temporal lobe structures, including the hippocampus, amygdala, and entorhinal cortex. Cytoarchitecture and cell densities are also reportedly altered in limbic structures: reduced cell number and size have been observed in the hippocampus, parahippocampal gyrus, and entorhinal cortex, along with disturbed cytoarchitecture involving cellular disarray in the hippocampus and the entorhinal and cingulate cortices. This distribution of structural abnormalities may help account for the symptoms of schizophrenia in that these limbic structures link neocortical association areas with the septum–hypothalamic complex and are therefore key in sensory information processing and sensory gating. Defects in this neural circuit could lead to a dissociation between neocortically mediated cognitive processing and limbic–hypothalamic emotional reactions and to a disturbed emotional experience of external sensory perceptions.
The structural alterations reported in schizophrenia are often subtle and certainly heterogeneous, contributing minimally to the elucidation of the disease if symptoms are to be attributed to defects in individual brain regions. Alternatively, it is currently hypothesized that the variety and severity of symptoms observed in schizophrenia can be explained by defects in several brain regions affecting the neural circuits that mediate higher cognitive functions. Such neural circuitry would include neocortical association areas, the limbic system and midline thalamic structures, with possible involvement of the basal ganglia.
The Neurodevelopmental Hypothesis of Schizophrenia
Several clinical observations point to neurodevelopmental abnormalities in schizophrenia: onset in adolescence in most patients and earlier onset among males, pronounced premorbid neurodevelopmental abnormalities, such as asociality and “soft” neurological signs, impaired cognitive and neuromotor functioning, minor physical anomalies, presence of structural abnormalities at the onset of the illness, which may predate the illness, and the absence of neurodegenerative processes.
Postmortem morphological observations in the brains of schizophrenic patients suggest that multiple aspects of neural development may be disrupted. The spatial disorganization of neurons found in the hippocampus and the inappropriate laminar location of cells in the entorhinal cortex, for example, suggest a potential defect in neuronal migration. Other evidence indicates that the organization of synaptic connectivity is affected. The functional circuitry of the frontal cortex undergoes significant remodeling until adolescence, mediated by an NMDA receptor-dependent mechanism. The number of these receptors is reduced in the brain of schizophrenics. Still other abnormalities involve biochemical alterations of membrane phospholipids in the dorsolateral prefrontal region. These changes include a decrease in phosphomonoesters and an increase in phosphodiesters. Such biochemical changes in membrane phospholipids are known to occur during normal synaptic pruning in adolescence, but appear to be exaggerated in the dorsolateral prefrontal cortex of schizophrenics. These enhanced biochemical changes may lead to greater synaptic pruning in schizophrenics than in normal individuals and contribute to the reduction in prefrontal neuropil volume that accompanies the disease.
The precise timing of neurodevelopmental abnormalities in schizophrenia remains controversial. Some implicate a fixed neural lesion from early life interacting with normal neurodevelopmental events that take place much later. Others posit that schizophrenia is caused by a deviation from normal brain maturational processes during late adolescence, involving large-scale synaptic elimination or pruning in brain regions critical for cognitive development. Still others propose that early brain pathology acts as a risk factor rather than a sufficient cause so that its effects can only be understood in the light of the individual’s later exposure to other risk and protective factors.
Genetics Play a Role in Schizophrenia
As for autism, a substantial body of evidence indicates that schizophrenia is associated with both genetic and environmental factors. Findings pointing to a significant genetic contribution come from family, twin, and adoption studies. Evidence for environmental factors derives from studies of prenatal intrauterine environment, neonatal obstetric complications, and postnatal brain insults. Currently, it remains an open question whether these environmental factors play a necessary role in the neurodevelopmental abnormalities responsible for psychotic brain disorders or whether their role is additive or interactive with genetic contributions.
People with schizophrenia are more likely than others to have schizophrenic relatives. The most compelling evidence is the 50% concordance rate for monozygotic twins relative to 15% concordance for dizygotic twins. Neuroimaging studies of discordant monozygotic twins consistently indicate more prominent structural brain abnormalities in the ill twin. More importantly, a neuroimaging study demonstrated that schizophrenia is associated with dysfunction of a prefrontal-limbic network (Weinberger, Berman, Suddath, & Torrey, 1992). In this study, monozygotic twins discordant for schizophrenia underwent PET blood flow scans while they performed a task known to activate the dorsolateral prefrontal cortex in normal subjects. This assessment, known as the Wisconsin Card Sorting Task (WCST), measures abstract problem solving requiring attention and working memory (see Fig. 50.4). The participant sees four cards bearing designs that differ in color, form, and number of elements. The subject’s task is to sort the stack of cards into piles in front of the stimulus cards. The subject is told whether the sorting choice is correct or incorrect. The test progresses such that the correct solution is initially stimulus color. Once mastered, the correct sorting strategy is switched without warning to form, requiring subjects to inhibit classifying cards on the basis of color. In a final phase the correct solution again changes unexpectedly, now to the number of elements. Shifting response strategies is difficult for patients with frontal lobe dysfunction. Among affected schizophrenic twins, prefrontal activation correlates strongly with both left and right hippocampal volumes; a relationship that is not observed in their unaffected twins. In addition, functional connectivity between the hippocampus and prefrontal cortex as measured by fMRI is weaker in schizophrenic patients than controls. These data suggest that schizophrenia may result from dysfunction distributed across a neural network involving prefrontal cortical areas and limbic structures.
Molecular studies demonstrate that schizophrenia is a complex genetic disorder, implicating several possible susceptibility loci, including regions on chromosomes 1q, 6p, 8p, 13q, and 22q. Of genes mapped, the 22q11 gene [a common functional polymorphism of catechol-O-methyltransferase (COMT), which is a methylation enzyme that metabolizes released dopamine] has been a popular candidate because of the long-hypothesized role of dopamine in schizophrenia. Recent molecular imaging studies in humans have also established that schizophrenia is associated with alterations in the dopamine system and these neurotransmitter abnormalities are shared by individuals at genetic and clinical risk for developing the illness.
Sex Hormones May Influence Neurodevelopmental Disorders
There are gender differences in the occurrence of neurodevelopmental disorders. The gender ratio of incidence is skewed toward boys for a variety of developmental disabilities, including severe mental retardation (1.3 boys/1 girl), speech and language disorders (2.6/1), learning difficulties (2.2/1), dyslexia (4.3/1), schizophrenia (1.9/1), and autism (4/1). Why? Although the existence of sex differences in the area of language and learning disorders has been contested on the grounds that it is a result of teacher and clinician bias in identifying these disorders, research supports the existence of a significant gender difference in the incidence of these disorders. Moreover, gender differences are also seen in clear-cut phenomena such as complications of pregnancy and birth, which are more common in male infants, and these differences cannot be explained as a reflection of investigator or clinician bias. Several theories attempt to explain these gender differences.
One theory posits that male but not female fetuses invoke a form of “antigenic” response from the mother during pregnancy, being recognized by the immune system as “foreign.” The hostile environment thus created for the male fetus in utero would explain not only a higher incidence of developmental disorders among boys, but also the finding that later-born sons are increasingly likely to be affected (consistent with increased immune responsiveness on repeated exposure).
Another theory (termed the Geschwind theory, after the neurologist Norman Geschwind) proposed that the uneven gender ratios in developmental disorders are associated with exposure to some “male factor” (possibly androgen) during the last trimester of fetal development. This factor acts to slow cortical maturation in male fetuses, particularly in the left hemisphere, rendering them more susceptible to perturbation during the normal course of development. This exposure to a male hormone would explain the higher incidence of developmental disorders among boys. Conversely, faster central neurons system development in female fetuses would enable them to better withstand insult during late pregnancy and birth. This idea is supported by evidence that female infants appear to show better cognitive recovery than male infants from intracranial hemorrhage associated with prematurity.
Summary
This section examined two forms of developmental abnormalities: autism and schizophrenia. In both cases it is possible to link the behavioral manifestations of the disorder to anomalies in neurobiological systems and to identify a genetic component. In both disorders, too, affected individuals do not show localized damage in circumscribed brain regions, but show evidence of cellular disturbance early in brain development (i.e., during neuromigration), which gives rise to pervasive and dysfunctional reorganization of critical neural systems underlying complex behaviors. Emerging evidence suggests that similar mechanisms may characterize other developmental disabilities, including dyslexia, mental retardation, and attention deficit disorders. Current findings also support the existence of sex differences in brain development and organization, which may in turn differentially affect the response of the brain to early injury.
Pathological Manifestations of Cognitive Aging: Dementia
Dementia is a generic term that refers to debilitating deterioration in more than one domain of cognitive function. The involvement of multiple capacities distinguishes dementia from other disorders, such as amnesia and aphasia, that affect a single functional domain (memory and language, respectively). Approximately 50 disorders are known to cause dementia. Most of these are progressive in nature, increasing in severity over time. The age at onset and the rate of progression of symptoms differ dramatically among the major dementing disorders. Most have an insidious onset and develop slowly, sometimes over a period of many years. These include Alzheimer’s disease, Huntington’s disease, frontotemporal dementia, and the dementia observed in a subpopulation of patients with Parkinson’s disease. The rare neurodegenerative disorder Creutzfeldt–Jakob disease also develops insidiously, but is distinguished by a very rapid rate of progression, often spanning a year or less from the onset of dementia to death. Vascular dementia (or multi-infarct dementia) follows still another pattern of decline. Initial cognitive symptoms develop acutely, but in this case the clinical course typically proceeds in a step-wise fashion over many years, with periods of relative stability punctuated by abrupt deterioration. Age is a major risk factor for dementing illnesses, and the following sections focus on the most prevalent example; Alzheimer’s disease.
Alzheimer’s Disease Is the Most Common Cause of Dementia
In 1906, the German scientist Alois Alzheimer reported a case study of a woman in her late 40s who developed memory impairment and deficits in other mental faculties that progressively worsened in severity until her death in her early 50s. Subsequent neuropathological examination of this patient’s brain revealed grossly apparent and widespread neocortical atrophy that Alzheimer concluded was inconsistent with any previously described disease. Although the disorder that would come to bear his name was originally thought to be quite rare, Alzheimer’s disease is now recognized as the most common cause of dementia, accounting for an estimated 50% of all cases, and the fifth leading cause of death among those over 65 years of age. Current estimates are that more than 5 million people in the United States, and 26 million people worldwide, suffer from Alzheimer’s disease. The prevalence of the disease is tightly coupled to age and increases after the fourth decade of life. The disease affects about 12% of people over the age of 65, and by 85, as many as 40% are afflicted. Demographic trends indicate that the elderly comprise the fastest growing segment of the population, and as a consequence, the number of people suffering the devastating impact of Alzheimer’s disease is projected to nearly triple by 2050. Not surprisingly, efforts to understand the basis of this and related disorders are among the most active research areas in neuroscience.
Memory Impairment Is a Central Feature of Alzheimer’s Disease
Alzheimer’s original case study provided an accurate description of the clinical course for many patients with this disease. Impairment in establishing and maintaining memories for recent events is frequently the initial sign that brings the patient to the attention of health care professionals. At this early stage, memory for the remote past is relatively preserved, as are other forms of memory that do not require medial temporal lobe structures. Thus, the neuropsychological profile of the Alzheimer’s patient provides important clues about the neuropathological progression of this disorder, suggesting that the systems responsible for declarative memory are especially vulnerable. Neuroanatomical studies of the diagnostic hallmarks of Alzheimer’s disease (described in the next section) confirm this proposal. Progression of the disorder is marked by inexorable decline across multiple cognitive capacities. Although generally alert and responsive in early and middle phases of the disorder, Alzheimer’s patients often experience difficulty identifying the meaning of simple words, uses for common household objects, or the meaning of numbers. Other disturbances can include general confusion, agitation, delusions, social disinhibition, and paranoia. Language abilities can deteriorate progressively, and at the end stage of disease, patients are often mute and densely amnesic for both recent and more remote events. As might be predicted on the basis of this broad profile of impairment, the distribution of neuropathology in individuals dying with late-stage Alzheimer’s disease is extensive, involving widespread areas of the limbic system and association cortices, along with many subcortical brain regions, including the basal forebrain cholinergic system, striatum, thalamus, and cerebellum.
Amyloid-Containing Plaques and Neurofibrillary Tangles Are Diagnostic Hallmarks of Alzheimer’s Disease That Preferentially Target Memory-Related Brain Regions
There are two types of neuropathological lesions that, when present in sufficiently high numbers, are the diagnostic hallmarks of Alzheimer’s disease (Fig. 43.6). Plaques comprise extracellular deposits of amyloid β-protein (Aβ) that, in their mature form, are typically surrounded by a shell of dystrophic neurites (see Fig. 43.6B). The neuritic component of plaques consists of morphologically abnormal dendrites and axons of multiple neuronal types, typically associated with substantial numbers of glial cells. The amyloid core of plaques can contain multiple species of Aβ, but prominently includes a form ending at amino acid 42 that is prone to aggregation (Aβ42), and a slightly shorter species (Aβ40) that is normally produced more abundantly in neurons. Plaques vary substantially in size, and although the time it takes for mature neuritic plaques to form is not known precisely, they are likely to evolve slowly, over many years.
Figure 43.6 Photomicrographs of neurofibrillary tangles (A) and amyloid-containing plaques (B) in the hippocampal formation of a patient that died with late-stage Alzheimer’s disease. Note that the size and morphological characteristics of plaques in B vary widely. Scale bar: 50 μm. Courtesy of P. Hof, Mount Sinai School of Medicine.
In contrast to extracellular plaques, the second neuropathological hallmark of Alzheimer’s disease is localized intracellularly. Termed neurofibrillary tangles, these are composed of densely packed, abnormal fibers that occupy much of the cytoplasm and proximal neuronal processes of affected cells (Fig. 43.6A). Indeed the density of these fibers in many tangle-bearing neurons is sufficiently high that the cell body appears swollen and the nucleus displaced. The term “paired helical filaments” (PHF), sometimes used to describe this type of pathology, derives from electron microscopic evidence that tangles consist of pairs of twisted filaments with a highly regular periodicity. Studies using biochemical and immunocytochemical methods have demonstrated that PHF are composed of a cytoskeletal protein found in normal, healthy neurons: the microtuble-associated protein tau. Cytoskeletal abnormalities can disrupt intracellular trafficking of materials that are critical for cell viability, culminating in neuron death. Defining the mechanisms that initiate the hyperphosphorylation of tau and its aggregation into mature neurofibrillary pathology is a prominent avenue of research on Alzheimer’s disease.
Investigations aimed at mapping the neuroanatomical distribution of pathology during early and later stages of Alzheimer’s disease help account for the progression of clinical symptoms in patients with this disorder. Early in the course of the disease, when memory impairment is often the predominant complaint, plaques and tangles are particularly abundant in the entorhinal cortex and associated projection targets in the hippocampus. Indeed, in many cases virtually all of the neurons that originate the perforant path projection to the hippocampus exhibit neurofibrillary tangles. Emerging evidence also suggests the balance of excitatory and inhibitory drive is disrupted in vulnerable circuitry early in the course of the disease, perhaps contributing to Aβ deposition. In this way the pathophysiology of Alzheimer’s disease appears to initially target cortical networks critical for normal memory. As the severity of cognitive impairment progresses, the distribution of neuropathology becomes more widespread, affecting cortical and subcortical systems that support language, semantic knowledge, abstract reasoning, and other capacities.
Genetic Contributions to Alzheimer’s Disease
Important clues about the basis of Alzheimer’s disease have come from the study of people afflicted with an “early onset” form of the disorder that emerges before the age of 65. This relatively rare and aggressive condition exhibits an increased prevalence in certain families and is therefore thought to have a significant genetic component. To date, three gene mutations linked to early onset Alzheimer’s have been identified: one involving the amyloid precursor protein gene on chromosome 21, another in the presenilin 1 gene on chromosome 14, and the third in the presenilin 2 gene on chromosome 1. Each of these mutations is expressed in an autosomal-dominant fashion such that half the offspring of an affected parent develop the disease. Importantly, the common thread among the gene mutations that cause early onset Alzheimer’s disease is that each of them influences processing of the amyloid precursor protein (reviewed in Selkoe, 2001).
Several additional lines of evidence support the suggestion that the deposition of amyloid is central to the pathogensis of Alzheimer’s disease. By far the most common form of the disorder (accounting for an estimated 98% of cases) has a relatively late onset, after age 65. Among a number of recently identified candidate genes, one risk factor for late-onset Alzheimer’s disease is the allelic composition of the apolipoprotein E gene on chromosome 19 (ApoE)—that is, a gene that codes for a glycoprotein involved in cholesterol transport and metabolism. The ApoE gene has three alleles (designated ApoE2, ApoE3, and ApoE4), and patients with Alzheimer’s disease carry the ApoE4 allele with a frequency greatly exceeding individuals without the disease. Moreover, the risk of disease is coupled to the number of copies of this specific allele: individuals with no copies of ApoE4 are less likely than the general population to develop Alzheimer’s disease, the presence of one ApoE4 allele increases the disease risk four times, and people with two copies of ApoE4 are eight times more likely than the population at large to develop Alzheimer’s. Although the precise mechanism by which ApoE4 mediates disease susceptibility is unknown, one hypothesis is that it involves the disruption of processes responsible for scavenging and clearing amyloid peptide from the extracelluar space in brain.
Additional findings relevant to the role of amyloid in Alzheimer’s disease come from research on Down’s syndrome. Down’s syndrome results from the presence of an extra copy of the same chromosome that carries the gene for the amyloid precursor protein; chromosome 21. Thus, it is of considerable interest that people with Down’s syndrome who survive until the fourth decade of life invariably develop a distribution of amyloid plaques in the brain similar to Alzheimer’s patients. To the degree that aberrant processing or clearance of amyloid comprises the common pathogenic mechanism of Alzheimer’s disease, current findings encourage the view that interventions targeting this pathway might realize an effective treatment for the most common cause of age-related dementia.
Prospects for the Treatment of Alzheimer’s Disease
Although Alzheimer’s disease is presently incurable, several pharmacological treatments have been approved for the clinical management of cognitive impairment associated with the disorder. All of these treatments target either acetylcholine or glutamate signaling, and while they offer modest, transient benefit for some Alzheimer’s patients, they fall far short of an effective cure or prevention. Preclinical progress toward the latter goal includes evidence that immunization with Aβ can both attenuate spatial learning impairment and blunt the progression of amyloid deposition in transgenic mouse models of Alzheimer’s disease (reviewed in Selkoe, 2001). Translating this approach to clinical practice faces substantial challenges, and initial attempts indicate that parallel treatment in people can induce life threatening cerebral inflammation. Nonetheless, the recent development of techniques for imaging brain amyloid deposition in vivo has generated considerable excitement, suggesting that it may soon be possible to identify at risk individuals very early in the course of disease, prior to the onset of cognitive decline, when intervention is most likely to be beneficial. The hopeful outlook based on these findings is that vaccine therapies might ultimately yield an effective route for the treatment and prevention of Alzheimer’s disease.
Summary
This chapter has considered the neurobiology of cognitive development and aging. Early cognitive development is marked by the emergence of a remarkably complex set of adaptive capacities, including language skills, reasoning abilities, attention, declarative memory, and executive functions. The vast majority of neurons—the essential building blocks of the central nervous system—are present at birth in humans, and rather than depending on the recruitment of new neurons, cognitive development appears to involve the fine tuning and establishment of appropriate circuitry among existing cells. Given the complexity of orchestration required, it is not surprising that many major disorders affecting cognitive function appear to result from a disruption of normal neurodevelopmental events. Higher order cognitive capacities are also vulnerable to deterioration during normal aging. Whereas widely distributed neuron death was previously presumed to underlie these deficits, it now appears that regionally selective changes in connectivity and cell biological function in critical circuits are the causative factors. Aging is also associated with some of the most devastating pathological conditions affecting cognitive function, including Alzheimer’s disease. The dramatic progress realized in recent years has, for the first time, established a basis for the rational development of strategies for disease prevention and treatment.
References
1. Alvarado MC, Bachevalier J. Revisiting the maturation of medial temporal lobe memory functions in primates. Learning & Memory. 2000;7:244–256.
2. Bachevalier J, Vargha-Khadem F. The primate hippocampus: Ontogeny, early insult and memory. Current Opinion in Neurobiology. 2005;15:168–174.
3. Baron-Cohen S, Ring HA, Wheelwright S, et al. Social intelligence in the normal and autistic brain: An fMRI study. The European Journal of Neuroscience. 1999;11:1891–1898.
4. Bauer PJ. Long-term recall memory: Behavioral and neuro-developmental changes in the first 2 years of life. Current Directions in Psychological Scienc. 2002;11:137–141.
5. Benes FM, Turtle M, Khan Y, Farol P. Myelination of a key relay zone in the hippocampal formation occurs in the human brain during childhood, adolescence, and adulthood. Archives of General Psychiatry. 1994;51:477–484.
6. Bizon JL, Gallagher M. More is less: Neurogenesis and age-related cognitive decline in Long-Evans rats. Science of Aging Knowledge Environment. 2005;7, re2.
7. Brody H. Structural changes in the aging nervous system. Interdisciplinary Topics in Gerontology. 1970;7:9–21.
8. Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nature Reviews Neuroscience. 2006;7:30–40.
9. Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. In: M. H. Johnson, ed. Brain development and cognition: A reader. Cambridge, MA: Blackwell; 1993:125–143.
10. Conel JL. The postnatal development of the human cerebral cortex. Vols. I–VI Cambridge, MA: Harvard University Press; 1939.
11. de Haan M, Mishkin M, Baldeweg T, Vargha-Khadem F. Human memory development and its dysfunction after early hippocampal injury. TINS. 2006;29:374–381.
12. Diamond A. The development and neural bases of memory functions as indexed by the AB and delayed response tasks in human infants and infant monkeys. Annals of the New York Academy of Sciences. 1990;608:637–676.
13. Fletcher, B. R., & Rapp, P. R. (2012). Normal neurocognitive aging. In Handbook of psychology: Biological psychology and neuroscience (2nd ed., Vol. 3). In press.
14. Geinisman Y, de Toledo-Morrell L, Morrell F, Persina IS, Rossi M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological disector technique. Hippocampus. 1992;2:347–444.
15. Giedd JN, Vaituzis AC, Hamburger SD, et al. Quantitative MRI of the temporal lobe, amygdala, and hippocampus in normal human development: Ages 4–18 years. Journal of Comparative Neurology. 1996;366:223–230.
16. Hedden T, Gabrieli JD. Healthy and pathological processes in adult development: New evidence from neuroimaging of the aging brain. Current Opinion in Neurology. 2005;18:740–747.
17. Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990;28:517–527.
18. Huttenlocher PR, De Courten C. The development of synapses in striate cortex of man. Human Neurobiology. 1987;6:1–9.
19. Klingberg T. White matter maturation and cognitive development during childhood. In: Nelson CA, Luciana M, eds. 2nd ed. Cambridge, MA: MIT Press; 2008;237–243. Handbook of developmental cognitive neuroscience Chap. 14.
20. Lenroot RK, Giedd JN. Brain development in children and adolescents: Insights from anatomical magnetic resonance imaging. Neuroscience and Biobehavioral Reviews. 2006;30:718–729.
21. Lenroot RK, Gogtay N, Greenstein DK, et al. Sexual dimorphism of brain development trajectories during childhood and adolescence, Neuroimage. 2007;36:1065–1073.
22. Lenroot RK, Schmitt JE, Ordaz SJ, et al. Differences in genetic and environmental influences on the human cerebral cortex associated with development during childhood and adolescence. Human Brain Mapping. 2009;30:163–174.
23. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419.
24. Ofen N, Kao Y-C, Sokol-Hessner P, Kim H, Whitfield-Gabrieli S, Gabrieli JDE. Development of the declarative memory system in the human brain. Nature Neuroscience. 2007;10:1198–1205.
25. Peters A, Morrison JH, Rosene DL, Hyman BT. Are neurons lost from the primate cerebral cortex during normal aging?. Cerebral Cortex. 1998;8:295–300.
26. Pelphrey K, Carter EJ. Brain mecahnisms for social perception: Lessons from autism and typical development. Annals of the New York Academy of Sciences, 1145 2008;283–299.
27. Rapp PR, Gallagher M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9926–9930.
28. Rapp PR, Deroche PS, Mao Y, Burwell RD. Neuron number in the parahippocampal region is preserved in aged rats with spatial learning deficits. Cerebral Cortex. 2002;12:1171–1179.
29. Raznahan A, Toro R, Daly E, et al. Cortical anatomy in autism spectrum disorder: an in vivo MRI study on the effect of age. Cerebral Cortex. 2010;20:1332–1340.
30. Raznahan A, Lee Y, Stidd R, et al. Longitudinally mapping the influence of sex and androgen signaling on the dynamics of human cortical maturation in adolescence. Proceedings of the National Academy of Sciences. 2010;107:16988–16993.
31. Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: Plasticity, network dynamics, and cognition. Progress in Neurobiology. 2003;69:143–179.
32. Schore AN. The experience-dependent maturation of a regulatory system in the orbital prefrontal cortex and the origin of developmental psychopathology. Development and Psychopathology. 1996;8:59–87.
33. Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiological Reviews. 2001;81:741–766.
34. Seress L. Pre- and postnatal morphological development of the human hippocampal formation. In: Nelson CA, Luciana M, eds. 2nd ed. Cambridge, MA: MIT Press; 2008;187–211. Handbook of developmental cognitive neuroscience Chap. 12.
35. Shaw P, Kabani NJ, Lerch JP, et al. Neurodevelopmental trajectories of the human cerebrtal cortex. The Journal of Neuroscience. 2008;28:3586–3594.
36. State MW. The genetics of child psychiatric disorders: Focus on autism and tourette syndrome. Neuron. 2010;68:254–269.
37. Wang M, Gamo NJ, Yang Y, et al. Neuronal basis of age-related working memory decline. Nature, 476 2011;210–213 http://dx.doi.org/10.1038/nature10243; 2011; [Epub ahead of print].
38. Weinberger DR, Berman KF, Suddath R, Torrey EF. Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: A magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. The American Journal of Psychiatry. 1992;149:890–897.
39. Wilson IA, Gallagher M, Eichenbaum H, Tanila H. Neurocognitive aging: Prior memories hinder new hippocampal encoding. TINS. 2006;29:662–670.
40. Zeamer A, Bachevalier J. Developmental trajectory of object recognition memory in infant rhesus monkeys with and without neonatal hippocampal lesions. The Journal of Neuroscience. 2010;30:9157–9165.
Suggested Readings
1. Bearden CE, Meyer SE, Loewy RL, Niendam TA, Cannon TD. The neurodevelopmental model of schizophrenia: Updated. In: Cicchetti D, Cohen DJ, eds. 2nd ed. New York, NY: John Wiley & Sons; 2006:542–569. Developmental psychopathology. vol. 3.
2. Burke SN, Barnes CA. Senescent synapses and hippocampal circuit dynamics. TINS. 2010;33:153–161.
3. Casey BJ, Tottenham N, Liston C, Durston S. Imaging the developing brain: What have we learned about cognitive development?. TRENDS in Cognitive Sciences. 2005;9:104–110.
4. Dawson G, Toth K. Autism spectrum disorders. In: Cicchetti D, Cohen DJ, eds. 2nd ed. New York, NY: John Wiley & Sons; 2006:318–357. Developmental psychopathology. vol. 3.
5. Dean RL, Bartus RT. Behavioral models of aging in nonhuman primates. In: Iversen LL, Iversen SD, Snyder SH, eds. New York, NY: Plenum; 1988:352–392. Handbook of psychopharmacology. Vol. 20.
6. Deboysson-Bardies B, de Schonen S, Jusczyk P, McNeilage P, Morton J. Developmental neurocognition: Speech and face processing in the first year of life Boston: Kluwer Academic; 1993.
7. Gallagher M, Rapp PR. The use of animal models to study the effects of aging on cognition. Annual Review Psychology. 1997;48:339–370.
8. Hernandez DG, Nalls MA, Gibbs JR, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Human Molecular Genetics. 2011;20:1164–1172.
9. Keshavan MS, Murray RM. Neurodevelopment and adult psychopathology Cambridge: Cambridge Univ. Press; 1997.
10. Lenroot RK, Giedd JN. Annual research review: Developmental considerations of gene by environment interactions. Journal of Child Psychology and Psychiatry. 2011;52:429–441.
11. Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiology of Aging. 2011;32:1161–1180.
12. Nelson CA, Luciana M. Handbook of developmental cognitive neuroscience 2nd ed. Cambridge, MA: MIT Press; 2008.
13. Nickl-Jockschat T, Habel U, Maria Michel MT, et al. Brain structure anomalies in autism spectrum disorder—a meta-analysis of VBM studies using anatomic likelihood estimation. Human Brain Mapping 2011; http://dx.doi.org/10.1002/hbm.21299; 2011; [Epub ahead of print].
14. Paus T. Mapping brain maturation and cognitive development during adolescence. TRENDS in Cognitive Sciences. 2005;9:60–68.
15. Schmitz C, Rezaie P. The neuropathology of autism: Where do we stand?. Neuropathology and Applied Neurobiology. 2008;34:4–11.
16. Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nature Medicine. 2011;17:1060–1065.
17. Sherwood CC, Gordon AD, Allen JS, et al. Aging of the cerebral cortex differs between humans and chimpanzees. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13029–13034.
18. Stern Y. Cognitive reserve. Neuropsychologia. 2009;47:2015–2028.
19. Stromswold K. The cognitive and neural bases of language acquisition. In: Gazzaniga M, ed. Cambridge, MA: MIT Press; 1995;855–870. The cognitive neurosciences .
20. Yassa MA, Mattfeld AT, Stark SM, Stark CEL. Age-related memory deficits linked to circuit-specific disruptions in the hippocampus. In: Gazzaniga M, ed. 2011;8873–8878. Proceedings of the National Academy of Sciences of the United States of America, 108 .