Chapter II.3.4
In Vivo Assessment of Tissue Compatibility
Introduction
The goal of in vivo assessment of tissue compatibility of a biomaterial, prosthesis or medical device is to determine the biocompatibility (as a key component of safety) of the biomaterial, prosthesis or medical device in a biological environment. Indeed, biocompatibility is a critical element in the performance of an implanted medical device, and many complications of clinical devices derive from inadequate biocompatibility. Moreover, the concept of biocompatibility has evolved in recent years as biomaterials technology has become more sophisticated, particularly in permitting the biomaterial to effect complex, and potentially beneficial, interactions with the surrounding tissues (Hench and Pollak, 2002; Williams, 2008; and see Chapter II.3.2). Thus, the contemporary definition of biocompatibility focuses on the ability of a medical device to perform with an appropriate host response in a specific application, and biocompatibility assessment serves as a measurement of the magnitude and duration of the pathophysiologic mechanisms that determine the host response. From a practical perspective, the in vivo assessment of tissue compatibility of medical devices is carried out to determine that the device performs as intended, and presents no significant harm to the patient or user simulating clinical use. In this chapter, the term “medical device” will be used to describe biomaterials, prostheses, artificial organs, and other medical devices, and the terms “tissue compatibility assessment,” “biocompatibility assessment,” and “safety assessment” will be considered to be synonymous.
The in vitro assessment of tissue compatibility is necessary to establish the biocompatibility safety and function (efficacy) of a medical device and its components under conditions of intended use. Intended use conditions are also utilized to select in vitro tests and animal models that will provide essential information to determine biocompatibility.
Recently, extensive efforts have been made by government regulatory agencies, e.g. the US Food and Drug Administration (FDA), and standards organizations, e.g. ASTM International, the International Organization for Standardization (ISO), and US Pharmacopeia (USP), to provide procedures, protocols, guidelines, and standards that may be used in the in vivo assessment of the tissue compatibility of medical devices. This chapter draws heavily on the ISO 10993 standard, Biological Evaluation of Medical Devices, in presenting a systematic approach to the in vivo assessment of tissue compatibility of medical devices.
The nature and extent of risk due to inadequate biocompatibility may be conceptualized as shown in Figure II.3.4.1. The first consideration in the selection of biomaterials to be used in device design should be the chemical, toxicological, physical, electrical, morphological, and mechanical properties of the biomaterial(s) to fulfill the intended use. Relevant to the overall in vivo assessment of tissue compatibility of a biomaterial or device is knowledge of the chemical composition of the materials, and the nature, degree, frequency, and duration of exposure of the device and its constituents to the tissues in which it will be utilized. Table II.3.4.1 presents a list of biomaterial components and characteristics that may affect the overall biological responses of the medical device. The range of potential biological hazards is broad, and may include short-term effects, long-term effects or specific toxic effects, which should be considered for every material and medical device. However, this does not imply that testing for all potential hazards will be necessary or practical.
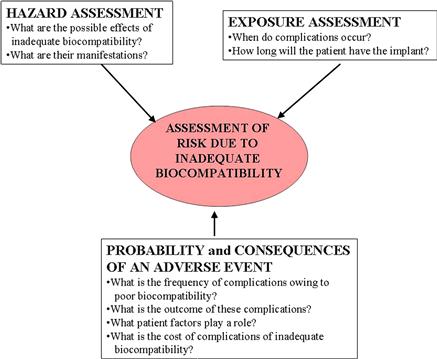
FIGURE II.3.4.1 The risks associated with inadequate biocompatibility are a composite function of the exposure (i.e., nature and duration), the hazard (i.e., effects on the recipient), and the probability and consequences of complications.
TABLE II.3.4.1 Biomaterials and Components Relevant to In Vivo Assessment of Tissue Compatibility
The material(s) of manufacture
Intended additives, process contaminants, and residues
Leachable substances
Degradation products
Other components and their interactions in the final product
The properties and characteristics of the final product
Selection of in Vivo Tests According to Intended Use
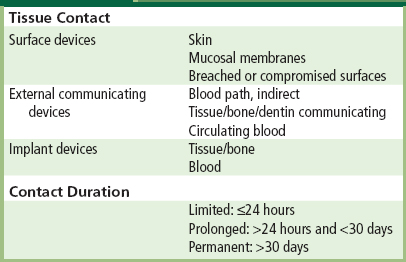
As it is recognized that biomaterials–tissue interaction may vary with anatomic site and duration of exposure, and application-specific conditions, in vivo tests for assessment of tissue compatibility are chosen to simulate end-use applications. To facilitate the selection of appropriate tests, medical devices with their component biomaterials can be categorized by the nature of tissue contact of the medical device, and by its duration of contact. Table II.3.4.2 presents medical device categorization by nature of tissue contact and contact duration. The tissue contact categories and subcategories, as well as the contact duration categories, have been derived from standards, protocols, and guidelines utilized in the past for safety evaluation of medical devices. Certain devices may fall into more than one category, in which case testing appropriate to each category should be considered.
TABLE II.3.4.2 Medical Device Categorization by Tissue Contact and Contact Duration
Biomaterial and Device Perspectives in IN Vivo Testing
Two perspectives may be considered in the in vivo assessment of tissue compatibility of biomaterials and medical devices. The first perspective involves the utilization of in vivo tests to determine the general biocompatibility of newly developed biomaterials for which some knowledge of the tissue compatibility is necessary for further research and development. In this type of situation, manufacturing and other processes necessary to the development of a final product, i.e., the medical device, have not been carried out. However, the in vivo assessment of tissue compatibility at this early stage of development can provide additional information relating to the proposed design criteria in the production of a medical device. While it is generally recommended that the identification and quantification of extractable chemical entities of a medical device should precede biological evaluation, it is quite common to carry out preliminary in vivo assessments to determine if there may be unknown or as yet identified chemical entities that produce adverse biological reactions. Utilized in this fashion, early in vivo assessment of the tissue compatibility of a biomaterial may provide insight into the biocompatibility, and thereby may permit further development of this biomaterial into a medical device. Obviously, problems observed at this stage of development would require further efforts to improve the biocompatibility of the biomaterial, and to identify the agents and mechanisms responsible for the adverse reactions. As the in vivo assessment of tissue compatibility of a biomaterial or medical device is focused on the end-use application, it must be appreciated that a biomaterial considered compatible for one application may not be compatible for another.
The second perspective regarding the in vivo assessment of tissue compatibility of medical devices focuses on the biocompatibility of the final product, that is, the fabricated medical device in the condition in which it is to be implanted. Thus, issues related to desired fabrications, interactions between biomaterials, and to mechanical conductors, etc., may come into play. Although medical devices in their final form and condition are commonly implanted in carefully-selected animal models to determine function, as well as biocompatibility, it may be inappropriate to carry out all of the recommended tests necessary for regulatory approval on the final device. In these situations, some tests may initially be carried out on biomaterial components of devices that have been prepared under manufacturing and sterilization conditions, and other processes utilized in the final product (see Table II.3.4.3).
TABLE II.3.4.3 In Vivo Tests for Tissue Compatibility
Sensitization
Irritation
Intracutaneous reactivity
Systemic toxicity (acute toxicity)
Subchronic toxicity (subacute toxicity)
Genotoxicity
Implantation
Hemocompatibility
Chronic toxicity
Carcinogenicity
Reproductive and developmental toxicity
Biodegradation
Immune responses
Specific Biological Properties Assessed by In Vivo Tests
In this section, brief perspectives on the general types of in vivo tests are presented. Details regarding these tests are found in the lists of standards provided at the end of this chapter. ISO 10993 standards advise that the biological evaluation of all medical device materials include testing for cytotoxicity, sensitization, and irritation. (Cytotoxicity tests are in vitro.) Beyond these fundamentals, the selection of further tests for in vivo biocompatibility assessment is based on the characteristics and end-use application of the device or biomaterial under consideration.
Sensitization, Irritation, and Intracutaneous (Intradermal) Reactivity
Exposure to, or contact with, even minute amounts of potential leachables from medical devices or biomaterials can result in allergic or sensitization reactions. Sensitization tests estimate the potential for contact sensitization to medical devices, materials, and/or their extracts. Symptoms of sensitization are often seen in skin, and tests are often carried out topically in guinea pigs. Test design should reflect the intended route (skin, eye, mucosa) and nature, degree, frequency, duration, and conditions of exposure of the biomaterial in its intended clinical use. While sensitization reactions are immune system responses to contact with chemical substances, ISO guidelines suggest irritation to be a local tissue inflammation response to chemicals, without a systemic immunological component. The most severely irritating chemical leachables may be discovered prior to in vivo studies by careful material characterization and in vitro cytotoxicity tests. Irritant tests emphasize utilization of extracts of the biomaterials to determine the irritant effects of potential leachables. Intracutaneous (intradermal) reactivity tests determine the localized reaction of tissue to intracutaneous injection of extracts of medical devices, biomaterials or prostheses in the final product form. Intracutaneous tests may be applicable where determination of irritation by dermal or mucosal tests is not appropriate. Albino rabbits are most commonly used.
Since these tests focus on determining the biological response of leachable constituents of biomaterials, their extracts in various solvents are utilized to prepare the injection solutions. Critical to the conduct of these tests is that the preparation of the test material and/or extract solution should be chosen to include testing for both water-soluble and fat-soluble leachables.
Systemic Toxicity: Acute, Subacute, and Subchronic Toxicity
Systemic toxicity tests estimate the potential harmful effects in vivo on target tissues and organs away from the point of contact (i.e., site of implantation) with either single or multiple exposures to medical devices, biomaterials, and/or their extracts. These tests evaluate the systemic toxicity potential of medical devices that release constituents into the body. These tests also include pyrogenicity testing, which assesses the induction of a systemic inflammatory response, often measured as fever.
In tests using extracts, the form and area of the material, the thickness, and the surface area to extraction vehicle volume are critical considerations in the testing protocol. Appropriate extraction vehicles, i.e., solvents, should be chosen to yield a maximum extraction of leachable materials for use in the testing. Mice, rats or rabbits are the usual animals of choice for the conduct of these tests, and oral, dermal, inhalation, intravenous, intraperitoneal or subcutaneous application of the test substance may be used, depending on the intended application of the biomaterial. Acute toxicity is considered to be the adverse effects that occur after administration of a single dose or multiple doses of a test sample given within 24 hours. Subacute toxicity (repeat dose toxicity) focuses on adverse effects occurring after administration of a single dose or multiple doses of a test sample per day during a period of from 14 to 28 days. Subchronic toxicity is considered to be the adverse effects occurring after administration of a single dose or multiple doses of a test sample per day given during a part of the lifespan, usually 90 days but not exceeding 10% of the lifespan of the animal.
Pyrogenicity tests are also included in the system toxicity category to detect material-mediated fever-causing reactions to extracts of medical devices or material. Although the rabbit pyrogen test has been the standard, the Limulus amebocyte lysate (LAL) reagent test has been used increasingly in recent years. It is noteworthy that no single test can differentiate pyrogenic reactions that are material mediated per se from those due to endotoxin contamination.
Genotoxicity
In vivo genotoxicity tests are carried out if indicated by the chemistry and/or composition of the biomaterial (see Table II.3.4.1) or if in vitro test results indicate potential genotoxicity (changes in deoxyribonucleic acid (DNA) that could lead to changes in cellular proliferation, differentiation, and/or function). Initially, at least three in vitro assays should be used, and two of these assays should utilize mammalian cells. The initial in vitro assays should cover the three levels of genotoxic effects: DNA destruction; gene mutations; and chromosomal aberrations (as assessed by cytogenetic analysis). In vivo genotoxicity tests include the micronucleus test, the in vivo mammalian bone marrow cytogenetic tests – chromosomal analysis, the rodent dominant lethal tests, the mammalian germ cell cytogenetic assay, the mouse spot test, and the mouse heritable translocation assay. Not all of the in vivo genotoxicity tests need be performed; the most common test is the rodent micronucleus test. Genotoxicitytests are performed with appropriate extracts or dissolved materials using appropriate media, as suggested by the known composition of the biomaterial.
Implantation
Implantation tests assess the local pathological effects on the structure and function of living tissue induced by a sample of a material or final product at the site where it is surgically implanted or placed into an implant site or tissue appropriate to the intended application of the biomaterial or medical device. In some cases, the anatomic site of implantation used for biocompatibility evaluation is not the same as the site of ultimate use, but has representative mechanisms and consequences of tissue–biomaterials interaction (e.g., subcutaneous implantation in rodents of bioprosthetic heart valve materials to study calcification that occurs as a major clinical limitation in humans; see Chapter II.4.5). The most basic evaluation of the local pathological effects is carried out at both the gross level and the microscopic level. Histological (microscopic) evaluation is used to characterize various biological response parameters (Table II.3.4.4). To address specific questions, more sophisticated studies may need to be done. Examples include immunohistochemical staining of histological sections to determine the types of cells present, and studies of collagen formation and destruction. For short-term implantation evaluation out to 12 weeks, mice, rats, guinea pigs or rabbits are the most common animals utilized in these studies. For longer-term testing in subcutaneous tissue, muscle or bone, animals such as rats, guinea pigs, rabbits, dogs, sheep, goats, pigs, and other animals with relatively long life expectancy are suitable. If a complete medical device is to be evaluated, larger species may be utilized so that human-sized devices may be used in the site of intended application. For example, substitute heart valves are usually tested as heart valve replacements in sheep, whereas calves are usually the animal of choice for ventricular assist devices and total artificial hearts.
TABLE II.3.4.4 Biological Response Parameters as Determined by Histological Assessment of Implants
Number and distribution of inflammatory cells as a function of distance from the material/tissue interface
Thickness and vascularity of fibrous capsule
Quality and quantity of tissue ingrowth (for porous materials)
Degeneration as determined by changes in tissue morphology
Presence of necrosis
Other parameters such as material debris, fatty infiltration, granuloma, dystrophic calcification, apoptosis, proliferation rate, biodegradation, thrombus formation, endothelialization, migration of biomaterials or degradation products
In all aspects of biocompatibility testing, it is important to recognize that the effects of the material on the surrounding tissues are generally superimposed on the events occurring during physiological wound repair induced by the surgery of implantation. This is particularly important in shorter-term experiments.
Hemocompatibility
Hemocompatibility tests evaluate effects on blood and/or blood components by blood-contacting medical devices or materials. In vivo hemocompatibility tests are usually designed to simulate the geometry, contact conditions, and flow dynamics of the device or material in its clinical application. From the ISO standards perspective, five test categories are indicated for hemocompatibility evaluation: thrombosis; coagulation; platelets; hematology; and immunology (complement and leukocytes). Two levels of evaluation are indicated: Level 1 (required); and Level 2 (optional). Regardless of blood contact duration, hemocompatibility testing is indicated for external communicating devices – blood path indirect; external communicating devices – circulating blood; and blood-contacting implant devices. Chapter II.5.4 gives further details on the testing of blood–material interactions.
Several issues are important in the selection of tests for hemocompatibility of medical devices or biomaterials. In particular, the hemocompatibility depends not only on the materials characteristics, but also on the fluid mechanics of the device (i.e., stasis promotes thrombus formation), and the coagulatability of the blood. Thus, in vivo testing in animals may be convenient, but anatomic differences among species and species-related differences in blood reactivity must be considered, and these may limit the predictability of any given test in the human clinical situation. While blood values and reactivity between humans and nonhuman primates are very similar, European Community law prohibits the use of nonhuman primates for blood compatibility and medical device testing. Hemocompatibility evaluation in animals is complicated by the lack of appropriate and adequate test materials, for example, appropriate antibodies for immunoassays. Use of human blood in hemocompatibility evaluation implies in vitro testing, which usually requires the use of anticoagulants that are not generally present with the device in the clinical situation, except for perhaps the earliest implantation period. Although species differences may complicate hemocompatibility evaluation, the utilization of animals in short- and long-term testing is considered to be appropriate for evaluating thrombosis and tissue interaction.
Chronic Toxicity
Chronic toxicity tests determine the effects of either single or multiple exposures to medical devices, materials, and/or their extracts during a period of at least 10% of the lifespan of the test animal, e.g., over 90 days in rats. Chronic toxicity tests may be considered an extension of subchronic (subacute) toxicity testing, and both may be evaluated in an appropriate experimental protocol or study.
Carcinogenicity
Carcinogenicity tests determine the tumorigenic potential of medical devices, materials, and/or their extracts from either single or multiple exposures or contacts over a period of the major portion of the lifespan of the test animal. Both carcinogenicity (tumorigenicity) and chronic toxicity may be studied in a single experimental study. With biomaterials, these studies focus on the potential for solid-state carcinogenicity, i.e., the Oppenheimer effect (see Chapter II.2.7). Thus, in carcinogenicity testing, controls of a comparable form and shape should be included; polyethyelene implants are a commonly used control material. The use of appropriate controls is imperative as animals may spontaneously develop tumors, and statistical comparison between the test biomaterial/device and the controls is necessary. To facilitate and reduce the time period for carcinogenicity testing of biomaterials, the FDA is exploring the use of transgenic mice carrying the human prototype c-Ha-ras gene as a bioassay model for rapid carcinogenicity testing.
Since tumors associated with clinical medical devices have been rare (see Chapter II.4.5) carcinogenicity tests should be conducted only if data from other sources suggest a tendency for tumor induction. However, considerations of carcinogenicity may become important in some future applications in which pleuripotential stem cells produced by any methodology are used.
Reproductive and Developmental Toxicity
These tests evaluate the potential effects of medical devices, materials, and/or their extracts on reproductive function, embryonic development (teratogenicity), and prenatal and early postnatal development. The application site of the device must be considered, and tests and/or bioassays should only be conducted when the device has a potential impact on the reproductive potential of the subject.
Biodegradation
Biodegradation tests determine the effects of a biodegradable material and its biodegradation products on the tissue response. They focus on the amount of degradation during a given period of time (the kinetics of biodegradation), the nature of the degradation products, the origin of the degradation products (e.g., impurities, additives, corrosion products, bulk polymer), and the qualitative and quantitative assessment of degradation products and leachables in adjacent tissues and in distant organs. The biodegradation of biomaterials may occur through a wide variety of mechanisms, which in part are biomaterial dependent, and all pertinent mechanisms related to the device and the end-use application of the device must be considered. Test materials comparable to degradation products may be prepared and studied to determine the biological response of degradation products anticipated in long-term implants. An example of this approach is the study of metallic and polymeric wear particles that may be present with long-term orthopedic joint prostheses. Moreover, for intentionally biodegradable scaffolds used in tissue engineering (see Chapter II.6.3), biodegradation and the attendant tissue response are important to follow histologically. Further insights on biodegradation are available in Chapters II.6.2 and II.6.3.
Immune Responses
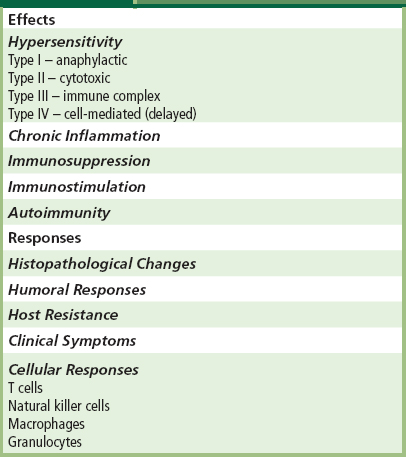
Immune response evaluation is not a component of the standards currently available for in vivo tissue compatibility assessment. However, ASTM, ISO, and the FDA currently have working groups developing guidance documents for immune response evaluation where pertinent. Synthetic materials are not generally immunotoxic (see Chapter II.2.3). However, immune response evaluation is necessary with modified natural tissue implants such as collagen, which has been utilized in a number of different types of implants and may elicit immunological responses. The Center for Devices and Radiological Health of the FDA has released a draft immunotoxicity testing guidance document (Langone, 1998) whose purpose is to provide a systematic approach for evaluating potential adverse immunological effects of medical devices and constituent materials. Immunotoxicity is any adverse effect on the function or structure of the immune system or other systems as a result of an immune system dysfunction. Adverse or immunotoxic effects occur when humoral or cellular immunity needed by the host to defend itself against infections or neoplastic disease (immunosuppression) or unnecessary tissue damage (chronic inflammation, hypersensitivity or autoimmunity) is compromised. Potential immunological effects and responses that may be associated with one or more of these effects are presented in Table II.3.4.5.
TABLE II.3.4.5 Potential Immunological Effects and Responses
Representative tests for the evaluation of immune responses are given in Table II.3.4.6. Table II.3.4.6 is not all-inclusive, and other tests that specifically consider possible immunotoxic effects potentially generated by a given device or its components may be applicable. Examples presented in Table II.3.4.6 are only representative of the large number of tests that are currently available. However, direct and indirect markers of immune responses may be validated and their predictive value documented, thus providing new tests for immunotoxicity in the future. Direct measures of immune system activity by functional assays are the most important types of tests for immunotoxicity. Functional assays are generally more important than tests for soluble mediators, which are more important than phenotyping. Signs of illness may be important in in vivo experiments, but symptoms may also have a significant role in studies of immune function in clinical trials and postmarket studies.
TABLE II.3.4.6 Representative Tests for the Evaluation of Immune Responses
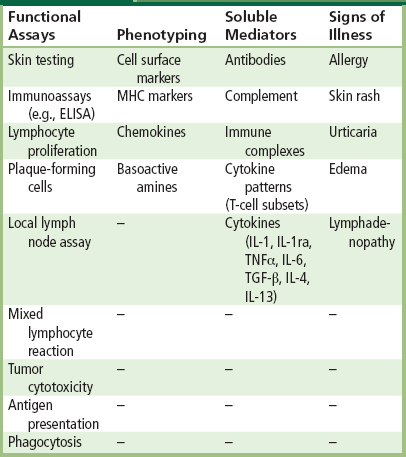
ELISA: Enzyme-linked immunosorbent assay; IL: Interleukin; TNF: Tumor necrosis factor; TGF: Transforming growth factor; MHC: Major histocompatibility complex.
Combination devices where drugs (or cells) are utilized within medical devices should also be considered for immune response evaluation. Hypersensitivity reactions have been reported with drug-eluting coronary stents (DES). With DES, concomitantly prescribed medications such as clopidogrel (platelet inhibitor) have been considered the causative agent for hypersensitivity, as well as the DES itself (Nebeker et al., 2006).
Selection of Animal Models for In Vivo Tests
Animal models are used to predict the clinical behavior, safety, and biocompatibility of medical devices in humans (Table II.3.4.7). The selection of animal models for the in vivo assessment of tissue compatibility must consider the advantages and disadvantages of the animal model for human clinical application. Several examples follow, which exemplify the advantages and disadvantages of animal models in predicting clinical behavior in humans.
Preclinical testing in animal models is an important part of the regulatory process, used to determine the safety and efficacy of devices prior to human clinical trials. The choice of the animal model and the selection of in vitro tests should be made according to the intended use of the respective medical device, prosthesis or biomaterial.
TABLE II.3.4.7 Animal Models for the In Vivo Assessment of Medical Devices
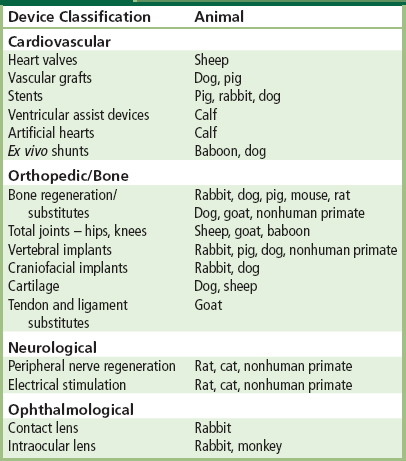
A single test animal may not assess all pertinent clinically-important complications. For example, as described earlier, sheep are commonly used for the evaluation of heart valves (see Chapters II.3.7 and II.4.5). This is based on size considerations, and also the propensity to calcify tissue components of bioprosthetic heart valves and thereby be a sensitive model for this complication. Thus, the choice of this animal model for bioprosthetic heart valve evaluation is made on the basis of accelerated calcification, the major clinical problem, assessed in rapidly growing animals which has its clinical correlation in young and adolescent humans. Nevertheless, normal sheep may not provide a sensitive assessment of the propensity of a valve to thrombosis, which may be potentiated by the reduced flow seen in abnormal subjects, but diminished by the specific coagulation profile of sheep.
The in vivo assessment of tissue responses to vascular graft materials is an example in which animal models present a particularly misleading picture of what generally occurs in humans. Virtually all animal models, including nonhuman primates, heal rapidly and completely with an endothelial blood-contacting surface. Humans, on the other hand, do not show extensive endothelialization of vascular graft materials, and the resultant pseudointima from the healing response in humans has potential thrombogenicity. Consequently, despite favorable results in animals, small-diameter vascular grafts (less than 4 mm in internal diameter) usually yield early thrombosis in humans, the major mechanism of failure which is secondary to the lack of endothelialization in the luminal surface healing response.
Originally, the porcine coronary artery model was considered the model of choice for the evaluation of arterial stents. More recently, the rabbit iliac artery model for the evaluation of drug-eluting stents has been considered to be more realistic, as endothelialization is slower in the rabbit model than in the porcine model, and inflammation is not as extensive in the rabbit (Nakazawa et al., 2008). Thus, endothelialization, healing, and inflammation in the rabbit iliac artery model may be closer to these responses in humans than the porcine coronary artery model.
The use of appropriate animal models is an important consideration in the safety evaluation of medical devices that may contain potential immunoreactive materials. The in vivo evaluation of recombinant human growth hormone in poly(lactic-co-glycolic acid)(PLGA) microspheres demonstrates the appropriate use of various animal models to evaluate biological responses and the potential for immunotoxicity. Utilizing biodegradable PLGA microspheres containing recombinant human growth hormone (rhGH), Cleland et al. (1997) used Rhesus monkeys, transgenic mice expressing hGH, and normal control (Balb/C) mice in their in vivo evaluation studies. Rhesus monkeys were utilized for serum assays in the pharmacokinetic studies of rhGH release, as well as tissue responses to the injected microcapsule formulation. Placebo injection sites were also utilized, and a comparison of the injection sites from rhGH PLGA microspheres and placebo PLGA microspheres demonstrated a normal inflammatory and wound-healing response with a normal focal foreign-body reaction. To further examine the tissue response, transgenic mice were utilized to assess the immunogenicity of the rhGH PLGA formulation. Transgenic mice expressing a heterologous protein have been previously used for assessing the immunogenicity of structural mutant proteins. With the transgenic animals, no detectable antibody response to rhGH was found. In contrast, the Balb/C control mice had a rapid onset of high-titer antibody response to the rhGH PLGA formulation. This study points out the appropriate utilization of animal models to not only evaluate biological responses, but also one type of immunotoxicity (immunogenicity).
Future Perspectives on In Vivo Medical Device Testing
As presented earlier in this chapter, the in vivo assessment of tissue compatibility of biomaterials and medical devices is dependent on the end-use application of the biomaterial or medical device. In this sense, the development and utilization of new biomaterials and medical devices will dictate the development of new test protocols and procedures for evaluating these new products. Furthermore, it must be understood that the in vivo assessment of tissue compatibility of biomaterials and medical devices is open-ended, and new end-use applications will require new tests.
The future development of medical devices is anticipated to provide more complexity to the composition and construction of these devices and, thus, to the array of potential biomaterials–tissue interactions. Thus, further studies will require a more sophisticated approach to test protocols and methodologies that must clearly identify biocompatibility and function. In this regard, new tests, methods, and animal models may have to be developed to adequately and appropriately characterize the biocompatibility and function of these new devices. Therefore, the development of guidelines and standards is dynamic and constantly evolving, driven by the complexity of new devices developed for application in tissue engineering, regenerative medicine, and nanomedicine.
Over the past half-century, medical devices and biomaterials have generally been “passive” in their tissue interactions. That is, a mechanistic approach to biomaterials–tissue interactions has rarely been used in the development of biomaterials or medical devices. Heparinized biomaterials are an exception to this, but considering the five subcategories of hemocompatibility, these approaches have minimal impact on the development of blood-compatible materials.
In the past decade, increased emphasis has been placed on bioactivity and tissue engineering in the development of biomaterials and medical devices for potential clinical application. Rather than a “passive” approach to tissue interactions, bioactive and tissue-engineered devices have focused on an “active” approach in which biological or tissue components, i.e., growth factors, cytokines, drugs, enzymes, proteins, extracellular matrix components, and cells that may or may not be genetically modified, are used in combinations with synthetic, i.e., passive, materials to produce devices that control or modulate a desired tissue response. Obviously, in vivo assessment of the targeted biological response of a tissue-engineered device will play a significant role in the research and development of that device, as well as in its safety assessment. It is clear that scientists working on the development of tissue-engineered devices will contribute significantly to the development of in vivo tests for biocompatibility assessment, as these tests will also be utilized to study the targeted biological responses in the research phase of the device development.
Regarding tissue-engineered devices, it must be appreciated that biological components may induce varied effects on tissue in the in vivo setting. For example, a simplistic view of the potentially complex problems that might result from a device releasing a growth factor to enhance cell proliferation is presented. The presence of a growth factor may result in markedly different cell proliferation, differentiation, protein synthesis, attachment, migration, shape change, etc., which would be cell-type dependent. Thus, different cell-type dependent responses in an implant site, reacting to the presence of a single exogenous growth factor, may result in inappropriate, inadequate or adverse tissue responses. These perspectives must be integrated into the planned program for in vivo assessment of tissue compatibility of tissue-engineered devices. Moreover, a major challenge to the in vivo assessment of tissue compatibility of tissue-engineered devices is the use of animal tissue components in the early phase of device development, whereas the ultimate goal is the utilization of human tissue components in the final device for end-use application. Novel and innovative approaches to the in vivo tissue compatibility of tissue-engineered devices must be developed to address these significant issues. Finally, the development of clinically useful tissue-engineered devices will require enhanced understanding of the influence of the patient and biomechanical factors on the structure and function of healed and remodeled tissues. It will also require new methodology for assessment of biocompatibility, and the dynamic progression of remodeling in vivo (Mendelson and Schoen, 2006).
Careful studies of retrieved implants to establish biomarkers and mechanisms of structural evolution will be critical (see Chapter II.1.5).
Bibliography
1. An YH, Friedman RJ. Animal Models in Orthopaedic Research. Boca Raton, FL: CRC Press; 1999.
2. Association for the Advancement of Medical Instrumentation. AAMI Standards and Recommended Practices, Vol 4. Biological Evaluation of Medical Devices, 1997. 1998;Vol. 4S.
3. Chapekar MS. Regulatory concerns in the development of biologic–biomaterial combinations. J Biomed Mater Res Appl Biomat. 1996;33:199–203.
4. Cleland JL, Duenas E, Daugherty A, Marian M, Yang J, et al. Recombinant human growth hormone poly(lactic-co-glycolic acid) (PLGA) microspheres provide a long lasting effect. J Control Release. 1997;49:193–205.
5. FDA (US Food and Drug Administration). Blue Book Memorandum G95–1: FDA-modified version of ISO 10993-Part 1, Biological Evaluation of Medical Devices – Part 1. Evaluation and Testing 1995.
6. Hench LL, Pollak JM. Third-generation biomedical materials. Biomaterials. 2002;295:1014–1017.
7. Langone JJ. Immunotoxicity Testing Guidance Draft Document, Office of Science and Technology. Center for Devices and Radiological Health, Food and Drug Administration 1998.
8. Nakazawa G, Finn AV, Ladich E, Ribichini F, Coleman L, et al. Drug-eluting stent safety: Findings from preclinical studies. Expert Rev Cardiovasc Ther. 2008;6(10):1379–1391.
9. Nebeker JR, Virmani R, Bennett CL, Hoffman JM, Samore MH, et al. Hypersensitivity cases associated with drug-eluting coronary stents. J Am Coll Cardiol. 2006;47:175–181.
10. Mendelson K, Schoen FJ. Heart valve tissue engineering: Concepts, approaches, progress, and challenges. Ann Biomed Eng. 2006;34:1799–1819.
11. Williams DF. On the mechanisms of biocompatibility. Biomaterials. 2008;29:2941–2953.
12. Yamamoto S, Urano K, Koizumi H, Wakana S, Hioki K, et al. Validation of transgenic mice carrying the human prototype c-Ha-ras gene as a bioassay model for rapid carcinogenicity testing. Environ Health Perspect. 1998;106(Suppl. 1):57–69.
ISO Standards
1. ISO 10993, Biological Evaluation of Medical Devices, International Standards Organization, Geneva, Switzerland.
2. ISO 10993-1 Evaluation and testing within a risk management system.
3. ISO 10993-2 Animal welfare requirements.
4. ISO 10993-3 Tests for genotoxicity, carcinogenicity, and reproductive toxicity.
5. ISO 10993-4 Selecton of tests for interactions with blood.
6. ISO 10993-5 Tests for in vitro cytotoxicity.
7. ISO 10993-6 Tests for local effects after implantation.
8. ISO 10993-7 Ethylene oxide sterilization residuals.
9. ISO 10993-9 Framework for the identification and quantification of potential degradation products.
10. ISO 10993-10 Tests for irritation and delayed-type hypersensitivity.
11. ISO 10993-11 Tests for systemic toxicity.
12. ISO 10993-12 Sample preparation and reference materials.
13. ISO 10993-13 Identification and quantification of degradation products from polymeric medical devices.
14. ISO 10993-14 Identification and quantification of degradation products from ceramics.
15. ISO 10993-15 Identification and quantification of degradation products from metals and alloys.
16. ISO 10993-16 Toxicokinetic study design for degradation products and leachables.
17. ISO 10993-17 Method for the establishment of allowable limits for leachable substances.
18. ISO 10993-18 Chemical characterization of materials.
19. ISO 10993-19 Physico-chemical morphological and topographical characterization of materials.
20. ISO-10993-20 Principles and methods for immunotoxicology testing of medical devices.
21. ISO 14971 Medical devices – application of risk management to medical devices.
ASTM International, 2008 Annual Book of ASTM Standards, Volume 13.01
1. F 895 Agar Diffusion Cell Culture Screening for Cytotoxicity.
2. F 2382 Assessment of Intravascular Medical Device Materials on Partial Thromboplastin Time (PTT).
3. E 1397 In Vitro Rat Hepatocyte DNA Repair Assay.
4. E 1398 In Vivo Rat Hepatocyte DNA Repair Assay.
5. F 1983 Assessment of Compatibility of Absorbable/Resorbable Biomaterials for Implant Applications.
6. F 981 Assessment of Compatibility of Biomaterials for Surgical Implants with Respect to Effect of Materials on Muscle and Bone.
7. F 756 Assessment of Hemolytic Properties of Materials.
8. F 1027 Assessment of Tissue and Cell Compatibility of Orofacial Prosthetic Materials and Devices.
9. F 1877 Characterization of Particles.
10. F 813 Direct Contact Cell Culture Evaluation of Materials for Medical Devices.
11. F 749 Evaluating Material Extracts by Intracutaneous Injection in the Rabbit.
12. F 750 Evaluating Material Extracts by Systemic Injection in the Rabbit.
13. F 2148 Evaluation of Delayed Contact Hypersensitivity Using the Murine Local Lymph Node Assay (LLNA).
14. F 1906 Evaluation of Immune Responses in Biocompatibility Testing Using ELISA Tests, Lymphocyte Proliferation, and Cell Migration.
15. F 619 Extraction of Medical Plastics.
16. F 2147 Guinea Pig: Split Adjuvant and Closed Patch Testing for Contact Allergens.
17. F 748 Selecting Generic Biological Test Methods for Materials and Devices.
18. F 1905 Selecting Tests for Determining the Propensity of Materials to Cause Immunotoxicity.
19. F 763 Short-Term Screening of Implant Materials.
20. F 1408 Subcutaneous Screening Test for Implant Materials.
21. F 719 Testing Biomaterials in Rabbits for Primary Skin Irritation.
22. F 1903 Testing for Biological Responses to Particles In Vitro.
23. F 720 Testing Guinea Pigs for Contact Allergens: Guinea Pig Maximization Test.
24. F 2065 Testing for Alternative Pathway Complement Activation in Serum by Solid Materials.
25. F 2567 Testing for Classical Pathway Complement Activation in Serum by Solid Materials.
26. F 1984 Testing for Whole Complement Activation in Serum by Solid Materials.
27. F 1904 Testing the Biological Responses to Particles In Vivo.
28. E 1263 Conduct of Micronucleus Assays in Mammalian Bone Marrow Erythrocytes.
29. E 1202 Development of Micronucleus Assay Standards.
30. E 1262 Performance of Chinese Hamster Ovary Cell/Hypoxanthine Guanine Phosphoribosyl Transferase Gene Mutation Assay.
31. F 1439 Performance of Lifetime Bioassay for the Tumorigenic Potential of Implant Materials.
32. E 1280 Performing the Mouse Lymphoma Assay for Mammalian Cell Mutagenicity.