X. SUMMARY OF THE STANDARD “TEXTBOOK” SYNDROMES OF UPPER AND LOWER MOTONEURON LESIONS AND THEIR VARIATIONS
A. Complete Table 7-7 to contrast the typical findings with a chronic upper motoneuron lesion and a chronic lower motoneuron lesion
TABLE 7-7 • Clinical Syndrome of UMN versus LMN Lesions
UMN |
LMN |
Characteristic |
Paralyzes movements in hemiplegic, quadriplegic, or paraplegic distribution, not in individual muscles |
||
Paralyzes individual muscles or sets of muscles in root or peripheral nerve distributions |
||
Atrophy of disuse only (late and slight) |
||
Atrophy of denervation (early and severe) |
||
Fasciculations and fibrillations |
||
Hyperactive MSRs |
||
Hypoactive or absent MSRs |
||
Clonus |
||
Clasp-knife spasticity |
||
Hypotonia |
||
Absent abdominal-cremasteric reflexes |
||
Extensor toe sign |
||
*Direct disease of the LMNs to the abdominal muscles abolishes these reflexes but disease confined to other LMNs does not. ABBREVIATIONS: LMN lower motoneuron; MSR muscle stretch reflex; UMN upper motoneuron. |
||
UMN |
LMN |
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
|
✓ |
B. Clinical variations in the pyramidal (upper motoneuron) syndrome
The level of the lesion along the course of the pyramidal tract (corticospinal and corticobulbar tracts; Fig. 2-29) determines the distribution of the weakness and other UMN signs. For the effect on the gait, see the gait essay at the end of Chapter 8.
1. Standard hemiplegic distribution: Unilateral interruption of the pyramidal tract at any level from the cortex to the pontomedullary junction causes weakness of the contralateral side of the body, from the lower facial muscles on down (Fig. 6-8B).
a. The degree of paralysis depends on how many of the axons of the pyramidal tract the lesion interrupts, and the individual variation in the number of crossed and uncrossed axons. In order to estimate the number of axons that will comprise the corticospinal tract, axon counts within each pyramid of the medulla have been performed and the number one million was eventually “accepted” (DeMyer, 1959). Yet, questions of prior histological technique suggest that this number may be overestimated and the actual number significantly lower, 50 to 70 000 (Wada et al, 2001).
Unique and pathognomonic neurological presentations can arise from small lacunar infarcts that occur within the deep areas of the cerebrum and brainstem and arise from occlusion of small penetrating arteries (Bailey et al, 2012; Fisher, 1982). One of these, pure motor hemiparesis, results from a lacunar infarction within the internal capsule, corona radiata, basis pontis and medulla oblongata, affecting the corticospinal tract within those locations and the topographic distribution of those fibers may result in variable weakness of the body (Melo et al, 1992; Jang, 2011).
b. The gradient and distribution of weakness are highly characteristic, essentially pathognomonic, of pyramidal tract interruption at cerebral, midbrain, and pontine levels. The weakness or paralysis involves the contralateral lower facial muscles and the contralateral extremities. While medullary level lesions usually spare the face, the course of some of these cortical-facial projections may not cross the midline until they have descended to the level of the medulla and give rise to patterns of facial weakness with lesions at that level (Urban et al, 2001) and present with facial weakness on the same side as the body (Claus et al, 1998).
In the extremities, the distal muscles are more affected than the proximal, and the finger movements suffer most of all. Unilateral paralysis of the tongue, palate, pharynx, and larynx is unusual, but the contralateral sternocleidomastoid is affected (Chapter 6). The opposite hemidiaphragm is weak during volitional breathing but acts bilaterally during automatic breathing (Chapter 6). UMN interruption equally paralyzes flexor and extensor muscles, contrary to traditional opinion (Thijs et al, 1998), but may reflect an intrinsic difference in strength as flexor muscle groups are stronger in the arms and extensors in the legs.
2. Monoplegia: A small lesion of the motor cortex or internal capsule, where the pyramidal fibers have a discrete somatotopic arrangement, may cause contralateral weakness of one arm or one leg or, rarely, just one or a few finger movements (Kim, 2002; Takahashi et al, 2002). A lesion of the leg area of its projection fibers will cause a contralateral lower extremity monoplegia. Interruption of the pyramidal tract on one side of the thoracic spinal cord causes a monoplegia of the ipsilateral leg.
3. Double hemiplegia: The lesion interrupts both pyramidal tracts somewhere rostral to the pontomedullary junction. The lower part of the face and all four extremities are paralyzed bilaterally. The Pt also shows pseudobulbar palsy.
4. Spastic diplegia: Bilateral pre- or perinatal cerebral lesions may result in a bilateral pyramidal syndrome, but with the legs more affected than the arms, and the bulbar muscles relatively spared. This reverses the arm-leg gradient of the weakness of double hemiplegia. Spastic diplegics often display involuntary movements such as athetosis.
5. Pseudobulbar palsy: Bilateral interruption of the corticobulbar component of the pyramidal tract (or all of the tract, at cerebral or brainstem levels) causes bilateral weakness of the oropharyngeal muscles with dysphagia, dysarthria, and spastic dysphonia, but the Pt shows exaggerated smiling and crying (Work et al, 2011; Chapter 6).
6. Locked-in syndrome: Bilateral interruption of the pyramidal tracts in the basis pontis or cerebral peduncles causes UMN paralysis of all volitional movements except vertical eye movements. The Pts remain conscious and not demented but are “locked-in” to themselves by the paralysis. See Chapter 12.
7. Quadriplegia (tetraplegia): Bilateral interruption of the pyramidal tracts in the caudal medulla or cervical region spares the face but paralyzes the volitional movements of the trunk and all four extremities. The Pt loses volitional bladder and bowel control and volitional control of breathing. The lesion spares those corticobulbar fibers to the face and bulbar muscles that depart rostral to the medullary level (Jagiella and Sung, 1989).
8. Paraplegia: Bilateral interruption of the pyramidal tracts caudal to the cervical region spares the arms but causes paralysis of the legs, with loss of bladder and bowel control.
C. A warning about the presence or absence of muscle stretch reflexes in relation to upper and lower motoneuron lesions
1. Although the general rule that LMN lesions cause hyporeflexia or areflexia and UMN lesions cause hyperreflexia holds, you now know that, in the acute phase of UMN lesions, the MSRs may be temporarily absent.
2. In the Guillain-Barré syndrome(GBS), the Pt typically suffers an ascending flaccid paralysis, beginning in the lower extremities. Typically also the Pt has the expected areflexia. In some Pts with the axonal form of this neuropathy, the MSRs are preserved or even hyperactive (Yuki and Hirata, 1998). Thus the distinction between a central or peripheral paralysis, that is, between a neuropathy and a myelopathy, becomes difficult on clinical grounds alone. The EMG aids materially in the differential diagnosis by placing the lesion in the peripheral nervous system and in differentiating the demyelinating from axonal forms of the syndrome (Rinaldi, 2013)).
D. Review of paralysis and sensory deficits immediately after acute spinal cord transection
1. Spinal cord transection at the medullocervical junction or at segments C1 to C3 (Fig. 2-5) results in complete apnea, complete quadriplegia, and complete anesthesia caudal to the lesion. The Pt dies of hypoxia within minutes unless given artificial respiration. The blood pressure also drops because interruption of the reticulospinal tracts stops the down flow of vasoconstrictor tone to the preganglionic sympathetic neurons in the intermediolateral column of the spinal cord. The sensorimotor deficits affect only the trunk and legs if the lesion is caudal to T1 (paraplegia).
2. During the stage of spinal shock, the Pt loses all somatomotor and most visceromotor responses caudal to the level of the lesion. The Pt shows flaccid paralysis of somatic muscles and sphincters and flaccid (atonic) bladder and bowel paralysis, with incontinence. All superficial and deep reflexes and most autonomic reflexes are abolished. If any visceral-related function remains, it is usually anal sphincter tone.
3. Gradually the classic UMN signs of increased MSRs, spasticity, and extensor toe signs will appear, as will reflex emptying of the bladder and bowel. In contrast, slowly evolving spinal cord compression results in spastic paralysis from the start. The Pt does not go through a phase of spinal shock.
4. After the phase of spinal shock, the transected spinal cord, isolated from the brain, can mediate simple actions such as MSRs and flexor withdrawal reflexes. It can also mediate reflex sweating, piloerection, micturition, defecation, and ejaculation (although the Pt feels no sensation), but it cannot produce any voluntary movements, respiratory drive, or respiratory-related reflexes such as coughing, sneezing, and hiccoughing. These reflexes require coordination of bulbar and spinal muscles. In summary, the isolated spinal cord can reflexly twitch some muscles, sweat, ejaculate and reflexly eliminate urine and feces, but the isolated spinal cord cannot breathe or make voluntary movements. These actions require intact reticulospinal and corticospinal tracts from the brain.
E. Patient analysis
1. Medical history: This 61-year-old man awakened one morning with double vision and inability to move his right side. He noticed mild numbness and tingling of his right side. When he called to his wife, he noticed slurring of his speech. He had been diabetic and hypertensive for many years.
2. Physical findings: The Pt was conscious, cooperative, and intact mentally. His left eye turned down and out, and the pupil was dilated. He could not adduct it. He had mild dysarthria. He had severe weakness of the right extremities, including the lower part of his face on the right. His right extremities were flaccid and somewhat hyporeflexic. He had a flexor plantar response on the left but little response at all on the right. He responded somewhat less to pain and touch on the right side.
3. Lesion localization
a. Before continuing with the text, you may want to propose where and what the lesion is. It may help you to review the brainstem cross sections shown in Figs. 2-15 to 2-18.
b. In localizing a single lesion to explain the Pt’s findings, first assemble the data that require explanation. The flaccidity and weakness, in a hemiplegic distribution, indicates interruption of the pyramidal tract in the acute stage of shock. The slight sensory findings suggest some involvement of a sensory pathway. The pupillary dilation, down and out position of the left eye, and inability to adduct it indicates a III nerve palsy. Therefore the Ex should “think circuitry” by visualizing the course of the pyramidal tract to try to locate a site where it comes into anatomic relation with a somatosensory pathway and CrN III.
c. The association of an LMN III nerve palsy on the left and hemiplegia of the right extremities suggests a lesion in the basis of the  mesencephalon/ pons/ medulla on the right/ left side. (
mesencephalon/ pons/ medulla on the right/ left side. ( mesencephalon; left (Fig. 2-16))
mesencephalon; left (Fig. 2-16))
d. Slight involvement of the medial lemniscus on the left could explain the sensory deficit on the right and indicate extension of the lesion from the basis into the tegmentum.
e. Magnetic resonance imaging showed patchy lucencies in the midbrain tegmentum and an overt infarct in the midbrain basis, where CrN III runs through it medially. At the midbrain level, the pathway for somatic sensation via the medial lemniscus had crossed the midline. Review Fig. 2-18 to see how a midbrain lesion could explain the III nerve palsy, right-sided pyramidal tract signs, and right-sided sensory findings. The Pt had the classic localizing findings of a brainstem lesion: a cranial nerve palsy on one side, and long tract signs on the other (Fig. 6-19).
f. What normal findings in this Pt would indicate that the lesion for the most part spares the dorsal part of the midbrain? (Hint: Review Figs. 2-18 and 6-19 to see what structures would have caused clinical signs if they had been destroyed. Does he show signs of the rostral midbrain syndrome [Table 5-2], or cerebellar signs in the left extremities?)
_________
_________
4. Clinical course: After several days the MSRs on the right became hyperactive, and spasticity, clonus, and a classic extensor toe sign appeared. The Pt did not regain any useful movements of his arm or fingers and could only walk when he wore a brace to support his knee. The III nerve palsy improved, but he remained with diplopia. The sensory loss disappeared, indicating only a transient ischemia of the medial lemniscus.
5. Course of corticobulbar fibers to the facial nucleus
a. Because the Pt’s hemiparesis included his face, the corticobulbar axons must have been in the basis of the midbrain. Infarcts of the basis pontis typically affect the face. The much rarer infarcts of the basis of the medulla (the medullary pyramids) often spare the face. What do these facts tell you about how far down the brainstem the fibers to the face travel?
_________
_________
BIBLIOGRAPHY · The Pyramidal Tract
Bailey EL, Smith C, Sudlow CLM, Wardlaw JM. Pathology of lacunar ischemic stroke in humans—a systematic review. Brain Pathol. 2012;22:583–591.
Claus SP, Kappelle LJ, Ramos LMP, van Gijn J. Stroke vignette: infarction of the medullary pyramid with hemiparesis including the face. Cerebrovasc Dis. 1998;8:245.
DeMyer W. Number of axons and myelin sheaths in adult human medullary pyramids. Neurology. 1959;9:42–47.
Fisher CM. Lacunar strokes and infarcts: a review. Neurology. 1982;32:871–876.
Jagiella WM, Sung JH. Bilateral infarction of the medullary pyramids in humans. Neurology. 1989;39:21–24.
Jang SH. Somatotopic arrangement and location of the corticospinal tract in the brainstem of the human brain. Yonsei Med J. 2011;52:553–557.
Kim JS, Chung JP, Ha SW. Isolated weakness of the index finger due to small cortical infarction. Neurology. 2002;58:985–986.
Melo TP, Bogousslavsky J, van Melle G, Regli F. Pure motor stroke: a reappraisal. Neurology. 1992;42:789–798.
Rinaldi S. Update on Guillain-Barré syndrome. J Peripher Nerv Syst. 2013;18:99–112.
Ropper AJ, Fisher CM, Kleinman GM. Pyramidal infarction in the medulla: a cause of pure motor hemiplegia sparing the face. Neurology. 1979;29:91–95.
Takahashi N, Kawamura M, Araki S. Isolated hand palsy due to cortical infarction; motor hand area. Neurology. 2002;58:1412–1414.
Thijs RD, Notermans NC, Wokke JHJ, et al. Distribution of muscle weakness of central and peripheral origin. J Neurol Neurosurg Psychiatry. 1998;65:794–796.
Urban PP, Wicht S, Vucorevic G, et al. The course of corticofacial projections in the human brainstem. Brain. 2001;124:1866–1876.
Wada A, Goto J, Goto N, et al. Are there one million nerve fibers in the human medullary pyramid? Okajimas Folia Anat Jpn. 2001;77:221–224.
Work SS, Colamonico JA, Bradley WG, Kaye RE. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28:586–601.
Yuki N, Hirata K. Preserved tendon reflexes in Campylobacter neuropathy. Ann Neurol. 1998;43:546–547.
XI. THE CONCEPT OF DEFICIT AND RELEASE PHENOMENA AFTER LESIONS OF THE MOTOR PATHWAYS
Upon this gifted age, in its dark hour,
Rains from the sky a meteoric shower
Of facts… they lie unquestioned, uncombined,
Wisdom enough to leech us of our ill
Is daily spun, but there exists no loom
To weave it into fabric, …
A. The theory of deficit and release phenomena
By this time, the array of positive and negative effects of neurologic lesions may seem puzzling, but we can subsume them under a theory of deficit and release phenomena.
1. Deficit phenomena are sensorimotor functions that the Pt loses after a neurologic lesion, for example, loss of movement or loss of vision.
2. Release phenomena are sensorimotor functions that increase or first emerge after a neurologic lesion. The release phenomenon may consist of an exaggeration of a normal action, for example, hyperactive MSRs, or a new response, for example, the change in the behavior of the large toe from flexion to extension (Babinski sign).
B. Deficit and release phenomena after upper motoneuron (pyramidal) lesions
1. Review the components of the UMN syndrome (Table 7-7) and list the deficit and release phenomena seen in the classic or chronic stage.
a. Deficit phenomena:
_________
_________
b. Release phenomena:
_________
_________
2. The extensor toe sign would be classed as a deficit/ release phenomenon because
_________
_________ release. It is a new behavior or response not present before the UMN lesion and is unmasked or released by the UMN lesion.)
3. In the acute phase after pyramidal tract interruption, particularly after spinal cord transection, the Pt may show only deficit/ release and no deficit/ release phenomena. ( deficit; release)
C. Pathophysiology of release phenomena
1. Release phenomena appear because the lesion has interrupted connections that presumably inhibited or suppressed the overactive function and because some intact pathway positively drives the overactivity.
2. Whether interruption of the pyramidal tract alone accounts for all of the release phenomena of the UMN syndrome remains unclear, but the clinician will make few errors in localization by assuming that pyramidal tract interruption is a necessary and sufficient condition for the full UMN syndrome to appear. However, involvement of other sensory and motor pathways may condition the expression and degree of the various components of the UMN syndrome.
D. Deficit and release phenomena after lower motoneuron lesions
By a stretch of the imagination, we can extend the deficit-release concept to the peripheral nervous system and even the sensory pathways.
1. Deficit phenomena after LMN lesions consist of:
a. Paresis or paralysis of individual muscles in a segmental or peripheral nerve distribution.
b. Decreased or absent MSRs.
c. Denervation atrophy (early and severe atrophy).
2. Release phenomena after LMN lesions consist of:
a. Fasciculations.
b. Fibrillations.
3. Regarding fasciculations, the disease of the LMN “releases” the depolarization mechanism of its own neuronal membrane, allowing the motor unit to fire randomly. In the case of fibrillations, the denervation “releases” the depolarization mechanism of the individual muscle fibers, allowing them to fire randomly.
E. Deficit phenomena after interruption of autonomic motor axons
1. Paralysis and atony of smooth muscle, thus abolishing peristalsis, propulsion, and emptying.
2. Vasomotor paralysis, with vasodilation, orthostatic hypotension, and impotence.
3. Anhidrosis.
4. Trophic changes consisting of hair loss, atrophy of skin, and dystrophy of nails.
F. Autonomic release or irritative phenomena after peripheral nerve lesions
In certain instances, lesions of peripheral nerves (or spinal cord lesions) release autonomic signs consisting of hyperhidrosis or vasoconstriction, rather than anhidrosis and vasodilation (see causalgia in Chapter 10).
G. Deficit and release phenomena after lesions of the basal motor nuclei
See Section M, page 295.
XII. INVOLUNTARY MOVEMENT DISORDERS
A. Introduction to the concept of voluntary and involuntary movements and the notion of free will
1. We experience ourselves as having free will. This experience leads us to classify behaviors intuitively as voluntary and involuntary. But then we must puzzle over behaviors such as breathing, bladder and bowel emptying, and postural reflexes that straddle the voluntary–involuntary dichotomy. For example, you can freely will yourself to hold your breath for a period of time, but ultimately you simply have to breathe. You have no choice. The physiologic imperative to act (ie, to emit a certain behavior) overpowers the will. People with mental disorders often experience their behaviors and even their very thoughts as involuntary or directed by external forces. Thus, Pts come to the physician because they experience behaviors and thoughts that they cannot willfully control.
2. By virtue of operational definitions, we can identify certain movements and behaviors that we can agree to class as voluntary or involuntary.
B. Working definition of voluntary and involuntary movements (behaviors)
1. A voluntary movement is one that the standard normal person can start or stop at the person’s own command or an observer’s command.
2. An involuntary movement is one that the standard normal person does not start and cannot stop at the person’s own or an observer’s command.
a. As strictly construed by neurologists, involuntary movements mean those patterns of muscle contractions (tremors and other movement sequences) caused by identifiable structural or biochemical lesions in the circuitry of the basal motor nuclei, reticular formation, and cerebellum.
b. Broadly construed, the concept of involuntary movements can also include the gamut of muscle fiber contractions of peripheral and central origins, extending from fibrillations to epileptic seizures.
3. Write out the definition of behavior given in Chapter 1, page 1:
_________
_________
4. Reread the statements in B1 to 2a and 2b above and substitute the word behavior for movement. It will give you a different feeling for the definition of behavior.
C. Clinical operations for identifying voluntary and involuntary movements
Neurologic lesions result in patterns of involuntary movements, recognizable from the history, inspection, and sometimes requiring laboratory tests, as for fibrillations. The operations for clinical analysis of involuntary movements follow.
1. Find out when the movements started, what conditions trigger or alleviate the movements, their relation to sleep and emotion, and their evolution over time. In other words, what is the history?
2. Describe the pattern of the movements, their distribution, rate, amplitude, and force. In other words, what are the physical findings?
3. Inspection is pivotal, allowing recognition without recourse to the Pt’s testimony, in most cases. To see is to diagnose, because most involuntary movements fall into stereotyped, identifiable patterns (Fig. 15-4).
D. Some normal involuntary movements
1. Physiologic synkinesia (syn = with; kinesis motion): A synkinesis is an involuntary or automatic movement that accompanies a voluntary movement. If you voluntarily close your eyes, your eyeballs automatically roll up (Bell phenomenon). Walk and your arms swing. Lean forward and your leg muscles automatically brace. Use these examples to identify other synkinesias (Hint: What about convergence of the eyes?). The degree of volition in the synkinesias varies. You cannot stop Bell phenomenon, but you can stop your arms from swinging when you walk.
2. Myoclonic jerks
a. Myoclonic jerks are sudden, brief, shock-like, involuntary twitches of individual muscles or sets of muscles. A myoclonic twitch may appear in a single muscle, such as the biceps, perhaps after unaccustomed work. Or a more widespread startle response may cause an upright jerk of the head as when the person is falling asleep. A sudden discharge in the reticular activating system of the brainstem causes the lightening, myoclonic jerk of the head and the sudden restoration of consciousness. Myoclonus may occur at rest or provoked by voluntary action (action myoclonus), or triggered by diverse stimuli (stimulus-sensitive or reflex myoclonus).
b. Epileptic myoclonic jerks, often refractory to treatment, occur in many CNS diseases. What is physiologic under one circumstance is pathologic under another.
c. Myoclonic jerks are separate from fasciculations. Define a fasciculation.
_________
_________
d. A twitch of the entire muscle or groups of muscles is called a _________
3. Benign fasciculations
a. Fasciculations appear in some normal persons, particularly after exercise. If the person has no weakness or other signs of LMN disease, the diagnosis is benign fasciculations. Twitching of an eyelid is an example.
b. What clinical and EMG findings would differentiate pathologic from benign fasciculations?
_________
_________
4. Physiologic tremor: See next section.
E. Tremors
1. Definition: Tremors are rhythmic, involuntary, oscillations of one or more regions of the body (Fig. 7-38).
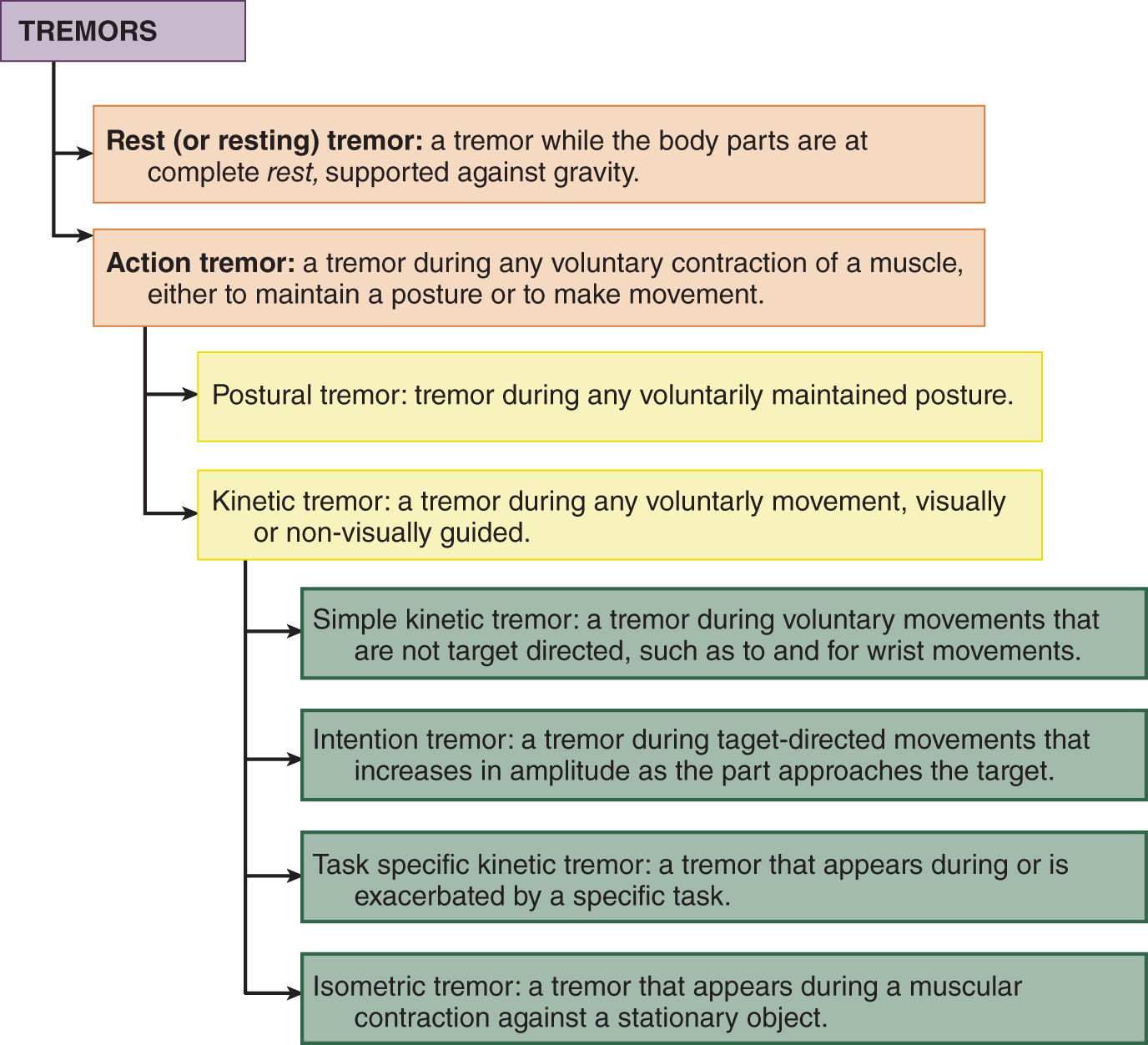
FIGURE 7-38. Classifications of tremors.
2. Clinical characteristics of tremors: Types of tremors differ in distribution, rate, and amplitude and whether they occur at rest or during voluntary muscular contractions. They also differ in response to drugs, in response to sleep and emotion, and in pathogenesis (Elias and Shah, 2014).
a. Distribution: Tremors most commonly affect the head, jaw, tongue, palate, and hands but may affect the trunk or legs.
b. Rate: Tremors vary in rate between 3 and 12 cps.
c. Amplitude: Tremors vary from barely perceptible to gross.
d. Relation to rest and volitional muscular contractions: Tremors dichotomize into two main groups: rest tremor and action tremors. Action tremors appear during voluntary muscular contractions, either to move a part or to maintain a voluntary posture. Action tremors include postural, kinetic, isometric, and task-specific types (Fig. 7-38).
e. Response of tremors to drugs: Drugs may increase or decrease tremor. Tremors commonly accompany lithium treatment. Anticholinergic drugs or dopamine decrease the tremor of Parkinson disease. Alcohol and propranolol decrease physiologic or essential tremor, whereas adrenalin increases them. Tremors, “the shakes,” commonly follow withdrawal from alcohol and other drugs.
f. Response of tremors to sleep and emotion: Like virtually all involuntary movements, tremors generally increase during emotional stress but dampen or disappear during tranquility and cease during sleep.
g. Lesion site for tremors: No single “tremorogenic” center exists. Tremors generally arise from disruption of the feedback circuits of the basal motor nuclei (Fig. 2-31), inferior olivary nucleus, and the cerebellum. Occasionally tremors occur with peripheral neuropathies.
3. Clinical characteristics of tremor types: Inspect the Pt for tremors under three conditions: rest, maintaining a posture, and during movement.
a. Rest (or resting) tremor is a tremor when the body parts are at complete rest, supported against gravity. Have the Pt sit with the arms relaxed and the forearms supported by the thighs or recline. Look for a tremor of the fingers and hands.
i. Rest tremor generally disappears during voluntary movement but increases during mental stress such as counting backward or when walking or moving another body part.
ii. The Ex can bring out a minimal or inapparent tremor or increase the amplitude of a rest tremor of the hands by having the Pt move the head from side to side (Froment maneuver).
iii. Rest tremor is highly characteristic of Parkinson disease.
b. Postural or position maintenance tremor occurs during the maintenance of any intentional posture, such as holding the head up, the trunk erect, or the arms out stretched. Postural tremor qualifies as an action tremor (Fig. 7-38) because the “action” refers to the sustained volitional contraction to hold the part in position. The hands, when extended in front, show a regular, rhythmic tremor of several cycles per second. If the Pt brings the finger in to touch his nose, intention tremor may appear. As the finger approaches the nose and the Pt attempts to stop the finger to maintain a position, the tremor reappears or heightens as an end point or terminal tremor. When the finger actually touches the nose, the tremor may or may not dampen. When the Pt returns his hands to his lap, at rest, any such action tremor disappears (Video 7-5).

Video 7-5. Unilateral postural tremor with dystonic features in a young patient with previous left cerebellar hemispheric and left thalamic infarcts.
c. Kinetic tremor or intention tremor (also called ataxic tremor) may appear during any voluntary movement. At rest, the hand remains still, but upon movement, as when the Pt does the finger-to-nose test, mild to moderate deviations detour the part from a straight line path. Intention and end-point tremor appear in various degrees and combinations. They implicate a lesion of the cerebellum or its efferent pathways.
d. Task-specific tremors appear during defined tasks, such as writing. An orthostatic tremor is a rare disorder of a very fine, rapid, 16-cps tremor of the legs appears mainly when the Pt stands, usually combined with a general feeling of unsteadiness or fear of falling and relieved by walking or leaning on nearby objects (Yaltho and Ondo, 2014).
e. Mixed tremors: A particular type of mixed tremor that appears at rest, while maintaining a posture, during voluntary movement and of low frequency (less than 5 Hz) with large and irregular amplitude often follows trauma to the midbrain or other midbrain lesions. Holmes tremor (thalamic, midbrain or rubral tremor) usually involves the proximal upper extremity unilaterally and with a variable delay between the lesion and onset. It is attributed to dysfunction within the nigrostriatal dopaminergic and cerebello-thalamic pathways (Video 7-6).

Video 7-6. Holmes tremor including palatal tremor.
Tremor is common in primary dystonia with estimates of 17% in those with late onset dystonia, both a postural and kinetic tremor, usually not present at rest, and typically (but not exclusively) involving the body part affected by dystonia (Defazio et al, 2013) and different in distribution from essential tremor suggesting they are different entities. A potential screening tool may be found in Pt handwritten spirals which appear to have a single predominant axis in essential tremor, lacking in dystonic tremor (Michalec et al, 2014).
F. Kymographic records of tremors
1. From these kymographic recordings, identify the type of tremor and the pathophysiologic basis:
a. The Pt is sitting quietly in a chair. An accelerometer attached to a hand records this tremor (Fig. 7-39).

FIGURE 7-39. Kymographic record of tremor.
b. The rate of the tremor is about ____ cps. (5 or 6)
c. Because the tremor occurs at rest but disappears during intentional movement, it qualifies as a rest/ intention/ postural tremor. ( rest)
d. This type of tremor signifies a lesion of the extrapyramidal pathways and is characteristic of the disorder called _________
2. The next Pt was sitting quietly, holding her arms extended in front, at shoulder level. Only a faint instability appeared. Figure 7-40 shows the tremor when the Pt attempted to touch her nose with her index finger. After she reached her nose, the tremor increased somewhat.
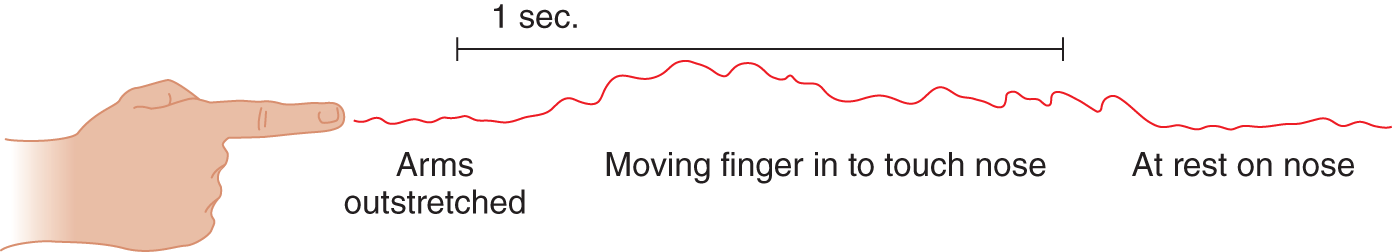
FIGURE 7-40. Kymographic record of tremor.
a. The tremor illustrated in Fig. 7-40 is called _________
b. It signifies a lesion of the pyramidal/ basal ganglia/ cerebellar pathways. ( cerebellar)
3. The next Pt had no tremor when sitting still, but when she held her arms straight out, a tremor appeared (Fig. 7-41).
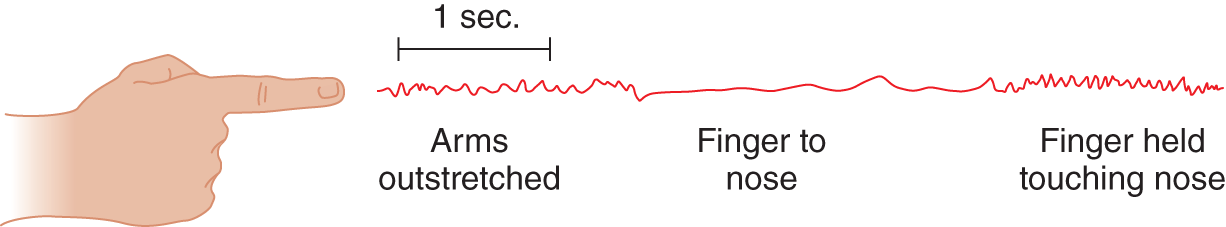
FIGURE 7-41. Kymographic record of tremor.
a. After she reached her nose, the tremor was accentuated. The tremor shown in the initial phase (Fig. 7-41), when the Pt was holding her arms extended and still, is called resting/ physiological/ postural tremor. ( postural)
b. It signifies a lesion of the _________
c. Because it may dampen during movement, postural tremor is like parkinsonian/ intention/ essential tremor. ( parkinsonian)
d. Give the full clinical characteristics of parkinsonian tremor.
_________
_________
G. Clinical features of several common tremor syndromes
1. Physiologic tremor
a. Self-demonstration of physiologic tremor: Insert a large sheet of paper between your index finger and the adjacent finger and hold your arm straight out in front of you. The rustling of the paper demonstrates physiologic tremor. The tremor has a frequency of about 10 cps.
b. Physiologic tremor is generally low in amplitude, relatively rapid (6-13 cps), and is most evident during movement or when a part sustains a posture. It varies from 6 cps in childhood, to 8 to 13 cps in adulthood, and back to around 6 cps in senility.
c. Physiologic tremor arises from a combination of neurally mediated oscillations and the ballistic effects of respiratory and cardiac actions (ballistocardiogram). Thus, it has neurologic and mechanical origins.
2. Emotional tremor, a normal phenomenon, to be distinguished from psychogenic tremor, is an enhanced physiologic tremor. It occurs at rest, but it worsens during volitional movement: Witness the quivering knees and quivering voice of the novice orator. From personal experience you know that emotional tremor is rapid/ very slow and of very great/ relatively low amplitude. ( rapid; relatively low)
3. Essential (familial) tremor
a. This autosomal dominant or “sporadic” disorder resembles physiologic tremor in frequency, with a range of 4 to 12 cps, but has a greater amplitude; despite its familial occurrence the genetics of essential tremor have yet to be completely defined (Kuhlenbäumer et al, 2014).
b. It affects the hands predominantly but may affect the head, bulbar muscles, and voice. It typically appears during a sustained posture but may appear during voluntary movements. It generally dampens or disappears with the Pt at rest. Tremors involving the tongue, trunk and lower limbs are rarely encountered (Kuhlenbäumer et al, 2014) (Video 7-7).

Video 7-7. Essential tremor involving head and voice tremor.
c. While the pathophysiological model for essential tremor suggested its origin within the pacemaking neurons in the inferior olivary nucleus (olivo-cerebellar output), currently it is believed to originate within the Purkinje cells and secondarily resulting in remodeling within the molecular and granular layers; may represent a neurodegenerative disease (Louis, 2014).
4. “Essential” versus symptomatic palatal tremor (previously referred to as palatal myoclonus).
a. Essential palatal tremor has the characteristics of essential tremor, but the Pt may experience an audible ear click and the tremor disappears during sleep. It is distinguished from the slow tremor (myorhythmia) of secondary palatal tremor by lack of associated neurologic deficits (eg, dysarthria, nystagmus and ataxia). Recently it was suggested that the essential type may consist of 3 overlapping variants: psychogenic where the tremor is variable, suppressible but occurs against the will, secondary that is partially volitional and temporarily suppressible and a volitional type where there is complete control and no disability and has been considered a “special skill” (Zadikoff et al, 2006; Stamelou et al, 2012; Biller and Espay 2013) (Video 7-8).

Video 7-8. Palatal tremor.
b. Symptomatic palatal tremor has a frequency of 110 to 160 beats/min, may affect other bulbar muscles derived from the branchial arches. It violates the law of disappearance of involuntary movements during sleep because it persists. It follows, with variable delays, lesions in the triangle between the contralateral dentate nucleus and ipsilateral red nucleus and inferior olivary nucleus (Guillain-Mollaret triangle). It is associated with hypertrophic olivary degeneration, a rare transsynaptic degeneration that results in enlargement rather than atrophy of the affected structure.
5. Parkinsonian tremor (Helmich et al, 2012)
a. Parkinsonian tremor has a frequency of 3 to 6 cps and low to moderate amplitude. Drum your fingertips on the table, timing 25 beats per 5 seconds, to observe two features of this tremor: moderate frequency and relatively low amplitude. It appears when the part is at rest, increases during mental and emotional tension, but disappears or dampens during intentional movement, and is absent during sleep. Often it appears asymmetrically. Typically affecting the hands and digits, the rustling of the thumb against the pads of the fingers resembles pill-rolling; hence its name, pill-rolling tremor. Voluntary head movements enhance the tremor (Froment maneuver).
b. A 4- to 8-cps postural tremor occurs also about as often as the classic rest tremor and at times distinguishing essential from Parkinson disease tremor can be difficult (Thenganatt and Louis, 2012).
c. Degeneration of the dopaminergic pathway that runs from the substantia nigra of the midbrain to the striatum causes Parkinson disease, but other neurons also degenerate. The parkinsonian triad of rest tremor, lead-pipe rigidity, and hypokinesia may appear as an entity or with associated widespread neuronal degeneration, known as the Parkinson plus syndromes (Bhidayasiri and Reichmann, 2013; Stameloua and Hoeglinger, 2013).
d. Parkinsonian tremor differs from most tremors by increasing/ decreasing during volitional movement, and resembles other involuntary movements by increasing/ decreasing during emotional stress and increasing/ disappearing during sleep. ( decreasing; increasing; disappearing)
e. The rest tremor of Parkinson disease, a hyperkinesia, contrasts with a reduction in the overall mobility of the Pt, called bradykinesia or hypokinesia. Paradoxically, an irresistible need to move, a physiologic imperative called akathisia (see Section XII P) also plagues the Pt, requiring abrupt, restless shifts of position against the background of bradykinesis and muscular rigidity. In a given Pt, one or more of the signs may predominate. Thus, one Pt displays mainly tremor and the next mainly rigidity. The basic motor signs of Parkinson disease include a quatrain of:
i. Rest tremor.
ii. Lead-pipe rigidity, often with a “cogwheeling,” ratchet-like yielding noted when checking muscle tone.
iii. Overall bradykinesia.
iv. Postural instability.
f. Derivative signs of the rigidity of the laryngeal muscles are loss of inflections of the voice, resulting in a characteristic monotonous tone, that is, plateau speech, and words running together (Walsh and Smith, 2012). Rigidity of the facial muscles results in an absence of emotional expression, that is, a masked face.
g. Oculogyric crises are spasms of upward deviation of the eyes or of the eyes and head. Common in post-encephalitic parkinsonism, oculogyric crises may occur in other disorders of the basal motor nuclei. Summarize for yourself the motor manifestations of parkinsonism.
h. Neurologic evaluation scales for following the course of parkinsonism document changes in Pt status (http://www.parkinson.org/Professionals/Professional-Resources/Screening-Instruments).
6. Neuropathic tremor: This tremor, usually an action type, occurs with a variety of acquired or hereditary peripheral neuropathies, more commonly with demyelinating than with axonal types (Saifee et al, 2013). The tremors are mainly postural and action tremors, but why some Pts develop tremor is not clear and there does not appear to be a clear relationship between severity of the neuropathy, proprioceptive loss, weakness or fatigue.
7. Drug-induced and toxic tremors: A number of tremors, as with hyperthyroidism, lithium treatment, delirium, and drug over dosage or withdrawal (including alcohol), share features of one or more of the foregoing types of tremors or come from an enhancement of physiologic tremor (Mehta et al, 2015). With the appearance of any new involuntary movement it is always prudent to review the patient’s medications as every type of abnormal movement has been described or attributed to a medication.
8. Psychogenic tremors have inconsistent, complicated patterns that change with circumstances (Thenganatt and Louis, 2012). Most of these tremors are action tremors. They often develop suddenly and sometimes have spontaneous remissions. The tremor decreases during distraction or volitional movement of the contralateral hand or stop briefly when testing motor tone. See Chapter 14 for other clinical features of psychogenic disorders.
H. Disorders to differentiate from tremors
1. Clonus is a repetitive MSR.
2. Asterixis consists of sudden lapses of a sustained posture, which may have a periodic or pseudo-rhythmic frequency. To elicit asterixis, ask the Pt to extend the arms straight out, with the wrists dorsiflexed. The wrist will then periodically drop and immediately re-extend (Video 7-9). First described in hepatic encephalopathy, it also appears in other metabolic-toxic encephalopathies and after structural lesions of the cerebello-thalamo-cortical circuits (Poewe and Djamshidian-Tehrani, 2015). The appearance of any new involuntary movement can indicate an underlying systemic disease that can resolve with treatment of the underlying disorder so, an early diagnosis is critical.

Video 7-9. Asterixis in a patient with traumatic skull base fracture.
3. Myoclonus
a. Myoclonus means shock-like, lightning fast contractions of parts of muscles or groups of muscles or their relaxation (eg, negative myoclonus). Individual movements are very brief but may be repetitive. They are irregular in rate and amplitude and symmetric or asymmetric (Lozsadi, 2012; Espay and Chen, 2013).
b. When restricted to one group of muscles, it is called focal or segmental myoclonus.
c. When widespread, the movements are multi-focal myoclonus or polymyoclonus.
d. Myoclonus appears in a large number of metabolic, toxic, and degenerative diseases and types of epilepsy and may arise from lesions at the spinal level, propriospinal myoclonus (van der Salm et al, 2014), subcortical or cortical levels (Carr, 2012).
e. The distinction between polymyoclonus, chorea, severe ataxia, hyperexplexia, and multiple tics is sometimes difficult.
f. A distinctive syndrome of opsoclonus myoclonus syndrome (dancing eye syndrome) affects children as an autoimmune disorder and as a distant effect of neuroblastoma. Personality changes are usual, particularly irritability (Hero and Schleiermacher, 2013).
4. Rhythmic myoclonus or Familial cortical myoclonic tremor and epilepsy (FCMTE). The Pt shows intermittent brief jerks, irregular or rhythmic, of slow frequency and often limited to segmental levels. The jerks are of high-frequency (7-18 cps) and epileptiform discharges often appear in the electroencephalogram (Licchetta et al, 2013).
5. Epilepsia partialis continua (of Kozhevnikov): The Pt shows continuous low-frequency jerks of one muscle group days and nights for weeks, months to years. The EEG shows focal epileptiform discharges and there are multiple etiologies and associated prognostic variables (Mameniskiene et al, 2011; Vein and van Emde Boas, 2011; Kravljanac et al, 2013). A chronic-progressive inflammatory syndrome is now considered a separate entity (Rasmussen syndrome or Rasmussen chronic encephalitis).
I. Review of tremors
Before proceeding with the text, review the tremor hyperkinesias by actually acting them out (Figs. 7-38 to 7-41). If you feel the need to, organize your own table or make a personal differential diagnostic dendrogram.
BIBLIOGRAPHY · Tremor and Parkinsonism
Bhidayasiri R, Reichmann H. Different diagnostic criteria for Parkinson disease: what are the pitfalls? J Neural Transm. 2013;120:619–625.
Biller J, Espay AJ. Nosography of the “essential”: volitional palatal tremor. Neurology. 2013;81:772–773.
Carr J. Classifying myoclonus: a riddle, wrapped in a mystery, inside an enigma. Parkinsonism Rel Disord. 2012;18(S1):S174–S176.
Defazio GH, Gigante AF, Abbruzzese g, et al. Tremor in primary adult-onset dystonia: prevalence and associated clinical features. J Neurol Neurosurg Psychiatry. 2013;84:404–408.
Elias WJ, Shah BB. Tremor. JAMA. 2014;311:948–954.
Espay AJ, Chen R. Myoclonus. Continuum (Minneap Minn). 2013;19:1264–1286.
Gajos A, Bogucki A, Schinwelski M, et al. The clinical and neuroimaging studies in Holmes tremor. Acta Neurol Scand. 2010;122:360–366.
Helmich RC, Hallett M, Deuschl G, et al. Cerebral causes and consequences of parkinsonian resting tremor: a tale of two circuits? Brain. 2012;135:3206–3226.
Hero B, Schleiermacher G. Update on Pediatric Opsoclonus Myoclonus Syndrome. Neuropediatrics. 2013;44:324–329.
Kravljanac R, Djuric M, Jovic N, et al. Etiology, clinical features and outcome of epilepsia partialis continua in cohort of 51 children. Epilepsy Res. 2013;104:112–117.
Kuhlenbäumer G, Hopfner F, Deuschl G. Genetics of essential tremor: meta-analysis and review. Neurology. 2014;82:1000–1007.
Licchetta L, Pippucci T, Bisulli F, et al. A novel pedigree with familial cortical myoclonic tremor and epilepsy (FCMTE): clinical characterization, refinement of the FCMTE2 locus, and confirmation of a founder haplotype. Epilepsia. 2013;54:1298–1306.
Louis ED. Essential tremor: from bedside to bench and back to bedside. Curr Opin Neurol. 2014;27:461–467.
Lozsadi D. Myoclonus: a pragmatic approach. Pract Neurol. 2012;12:215–224.
Mameniskiene R, Bast T, Bentes C, et al. Clinical course and variability of non-Rasmussen, nonstroke motor and sensory epilepsia partialis continua: a European survey and analysis of 65 cases. Epilepsia. 2011;52:1168–1176.
Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. 2015;33:153–174.
Michalec M, Hernandez N, Clark LN, Louis ED. The spiral axis as a clinical tool to distinguish essential tremor from dystonia cases. Parkinsonism Rel Disord. 2014;20:541–544.
Poewe W, Djamshidian-Tehrani A. Movement disorders in systemic disease. Neurol Clin. 2015;33:269–297.
Saifee TA, Schwingenschuh P, Reilly MM, et al. Tremor in inflammatory neuropathies. J Neurol Neurosurg Psychiatry. 2013;84:1282–1287.
Stamelou M, Saifee TA, Edwards MJ, Bhatia KP. Psychogenic palatal tremor may be under recognized: reappraisal of a large series of cases. Mov Disord. 2012;27:1164–1168.
Stameloua M, Hoeglinger GU. Atypical Parkinsonism: an update. Curr Opin Neurol. 2013;26:401–405.
Thenganatt MA, Louis ED. Distinguishing essential tremor from Parkinson’s disease: bedside tests and laboratory evaluations. Expert Rev Neurother. 2012;12: 687–696.
Van der Salm SMA, Erro R, Cordivari C, et al. Propriospinal myoclonus: clinical reappraisal and review of literature. Neurology. 2014;83:1862–1870.
Vein AA, van Emde Boas W. Kozhevnikov epilepsy: the disease and its eponym. Epilepsia. 2011;52:212–218.
Walsh B, Smith A. Basic parameters of articulatory movements and acoustics in individuals with Parkinson’s disease. Mov Disord. 2012;27:843–850.
Yaltho TC, Ondo WG. Orthostatic tremor: a review of 45 cases. Parkinsonism Rel Disord. 2014;20:723–725.
Zadikoff C, Lang AE, Klein C. The ‘essentials’ of essential palatal tremor: a reappraisal of the nosology. Brain. 2006;129:832–840.
J. Nontremor types of hyperkinesias
1. Several hyperkinesia display characteristic clinical features that predict a probable lesion site (Table 7-8).
TABLE 7-8 • Clinicopathologic Correlations Between Movement Disorders and Lesions of the Basal Motor Nuclei
Movement disorder |
Classic lesion site |
Chorea: multiple, quick, random movements (“the fidgets”) usually most severe in the appendicular muscles |
Striatum, atrophy, autoimmune disease; may also occur after lesions in a subthalamic red nucleus lesion |
Athetosis: slow, writhing movements most severe in the appendicular muscles |
Diffuse hypermyelination of the corpus striatum and thalamus, as in cerebral palsy |
Dystonia: long, sustained twisting movements most severe in axial muscles |
Genetic, acquired, or pharmacologic lesion of basal motor nuclei |
Hemiballismus: wild, flinging movements of half of the body |
Hemorrhagic lesion of contralateral subthalamic nucleus, usually in a hypertensive patient |
Parkinson Disease (paralysis agitans): pill-rolling rest tremor of 5-6 cycles/s of the fingers, lead-pipe rigidity, and akinesia |
Degeneration of the substantia nigra |
Rest, postural, and terminal tremor (Holmes or “rubral” tremor) |
Midbrain lesion, in the region of the red nucleus and superior cerebellar peduncle, often posttraumatic |
2. Chorea refers to incessant, random, moderately quick movements—a grimace, elevation of a finger or arm, a misstep when walking, an interruption when speaking. Overall, chorea resembles the “fidgets.” One part or another of the body is flickering into motion all of the time. The movements resemble a choreographer working out the movements for a dance or, perhaps better described, they simulate fragments of normal movements. For instance, at one time or another, after you start to make an inappropriate movement (perhaps reaching up to pick your nose), you may suddenly decide to arrest it midway. Or after starting such a movement, you may have diverted it to brushing back your hair. Patients with chorea may employ this ruse. Nevertheless, an observer can perceive the stoppage or the diversion in the continuity of the initial movement. The origin of chorea has yet to be determined, but appears to result in an imbalance between facilitation and inhibition during intended movements (Hallet and Obeso, 2015) (Video 7-10).

Video 7-10. Huntington chorea before and after treatment.
3. Athetosis refers to slow, writhing movements of the fingers and extremities. If severe, athetosis affects speech and some proximal movements. These movements wax and wane and generally do not hold the part in a fixed posture. Athetosis often accompanies or follows partial interruption of the pyramidal tracts, particularly in Pts with cerebral palsy and spastic quadriplegia or diplegia. The quick random fidgety movements, called _________
4. Dystonia refers to sustained or intermittent muscle contractions that cause abnormal, often repetitive, movements, postures, or both. They are typically patterned, twisting, and at times tremulous; often initiated or worsened by voluntary movement. The prolonged muscular contractions hold the part in one position for periods and may lead to pretzel-like body positions with fixed scoliosis and fixed contractures of joints.
a. Focal dystonia, such as spasmodic torticollis and writer’s cramp, may affect more or less restricted groups of muscles.
b. The sustained postural deviations of dystonia differ from the quick movements of chorea and the slower writhing, mainly distal movements called _________
c. Although dystonia is traditionally classed as an extrapyramidal movement disorder, the lesion site and pathophysiology of the hereditary form are unknown (Klein, 2014), but dystonia can result from known, acquired lesions of the basal motor nuclei and neuroimaging provides evidence that the basal ganglia and thalamocortical network are major determinants in its pathophysiology (Obeso et al, 2014; Stoessl et al, 2014) (Video 7-11).

Video 7-11. Focal dystonia of the left hand in a patient with prior aneurysmal SAH complicated by right hemispheric infarction.
5. Hemiballismus refers to violent flinging movements of one-half of the body. Ballista means to throw, as in ballistics. The Pt’s arm thrashes about as if it were trying to fling away a handful of snakes. Hemiballismus usually appears abruptly in elderly hypertensive Pts. The lesion is predictable. A peculiar, sharply delimited hemorrhage destroys the contralateral subthalamic nucleus of Luys or its immediate surrounding pathways. This almond-sized and almond-shaped diencephalic nucleus belongs to the basal motor nuclei. Hemiballismus may also result from lesions in the caudate, putamen, globus pallidus, precentral gyrus, or thalamic nuclei (Hawley and Weiner, 2012).
6. Tics are quick, lightning fast, stereotyped, involuntary movements of face, tongue, upper extremities, or phonations. (Martino and Mink, 2013). In contrast to the preceding hyperkinesias, the sequence of movements is identical each time if the Pt has one type of tic. However, the Pt may have multiple kinds of tics. All of us display minor tics: wrinkling of the forehead, followed by blinking the eyes, or hitching up the trousers, or a shrug of the shoulder. Athletes display a number of tic-like maneuvers, as when a basketball player prepares to shoot a free throw, or a tennis player prepares to serve. Tics increase during emotional stress, are less prominent during periods of concentration, and often abate during sleep. Although mostly of low amplitude, tics, when violent, may throw the Pt to the floor, thus resembling an exaggerated startle response called hyperekplexia. Of the movement disorders discussed, tics, being quick and usually of low to moderate amplitude, most closely resemble chorea/ athetosis/ hemiballismus. ( chorea)
7. Multiple tic syndrome of Gilles de la Tourette: Some tics are regarded as psychogenic, but they figure prominently in an organic disorder called Tourette syndrome, which has three major features:
a. Multiple tics that change from time to time.
b. Involuntary respiratory actions and vocalizations with squawks, barks, howls or sniffs, humming, and sometimes the involuntary utterance of expletives.
c. Personality traits: sometimes rigid, obsessive-compulsive, or abrasive and attention deficit hyperactivity disorder (Shprecher et al, 2014).
K. Continuum between the named types of hyperkinesias
Dystonia, athetosis, chorea, and tics represent way stations along a continuum of involuntary movements, not necessarily discrete entities. They differ perhaps more in their speed than in any other way. Tics and chorea are the fastest, with each movement measured in a second or even less. Next comes athetosis, lasting just a little longer, in seconds. Then comes dystonia, which lasts many seconds to minutes or even longer, as in spasmodic torticollis, a form of dystonia in which the head remains deviated for long periods.
Hemiballismus differs from chorea in its more violent amplitude and unilaterality. Hemichorea may occur, but it is most frequently bilateral, as in Huntington or Sydenham chorea.
L. Drug-induced extrapyramidal movement syndromes and the tardive dyskinesias
Various tranquilizers, antipsychotics, and antidepressant drugs as well as other medications can result in a wide spectrum of movement disorders (Mehta et al, 2015). These chemical lesions mimic the effect of anatomic lesions in producing hypo- and hyperkinesias, ranging from parkinsonism to dystonia. The involuntary movements often predominately affect facial, oral, buccal, and pharyngeal movements, with dysphonia and dysphagia (Meige syndrome). Unfortunately, tardive dyskinesia may be permanent and very resistant to therapy. Levodopa, used to treat parkinsonism, also produces dyskinesias.
M. The concept of deficit and release phenomena after lesions of the basal motor connections
For general discussion, see Section XI of this chapter.
1. Deficit phenomena after lesions of the basal motor circuitry include overall bradykinesia, gait impairment, masked facies, and loss of voice inflection.
2. Release phenomena include the hypertonia called lead-pipe rigidity, tremor (generally tremor of the part at rest), akathisia, and the patterned hyperkinesias such as tics, chorea, athetosis, dystonia, and hemiballismus. Whether the concept of release phenomena applies to these phenomena requires some stretch of the imagination. Even less does the concept apply to the other syndromes of excessive behavior, such as hyperactivity.
N. Rating scales for involuntary movement disorders
Various rating scales such as the Abnormal Involuntary Movement Scale enable the Ex to quantify various involuntary movements to determine the degree of disability, to follow the course of the disease, and to document the effects of treatment (Herndon, 2006).
O. Some simple, general tests for motor dysfunction: writing, finger-tapping speed, the Archimedes spiral, and activities of daily living
These tests are useful in analyzing most motor disorders, especially in Pts with hemiparesis, tremor, rigidity, or ataxia, not only for differential diagnosis but also to appreciate functional disability.
1. Writing: Watch the Pt write spontaneously and, if a motor disorder or cerebral disorder is suspected, write a sentence to dictation. Like the gait, most motor disorders affect writing. The ataxic dysgraphia of cerebellar disease, the micrographia of rigidity, and the tremulous dysgraphia of essential tremor stand out vividly from each other.
2. Finger-tapping speed
a. Technique: For a convenient bedside test in lieu of an actual counter (Finger Tapping Test or FTT; Strauss, 2006), place a deck of cards or if one can still be found an ordinary audiocassette on the table and have the Pt grasp it between the thumb and third digit, leaving the index finger free to tap (Fig. 7-42). Ask the Pt to tap at a comfortable rate to prevent fatigue, for several seconds (Arias et al, 2012). The Ex should demonstrate the test first. Normal subjects when asked to tap as rapidly as possible can do so at a rate of around 50 taps per 10 seconds. Children and the aged tap at slower rates.
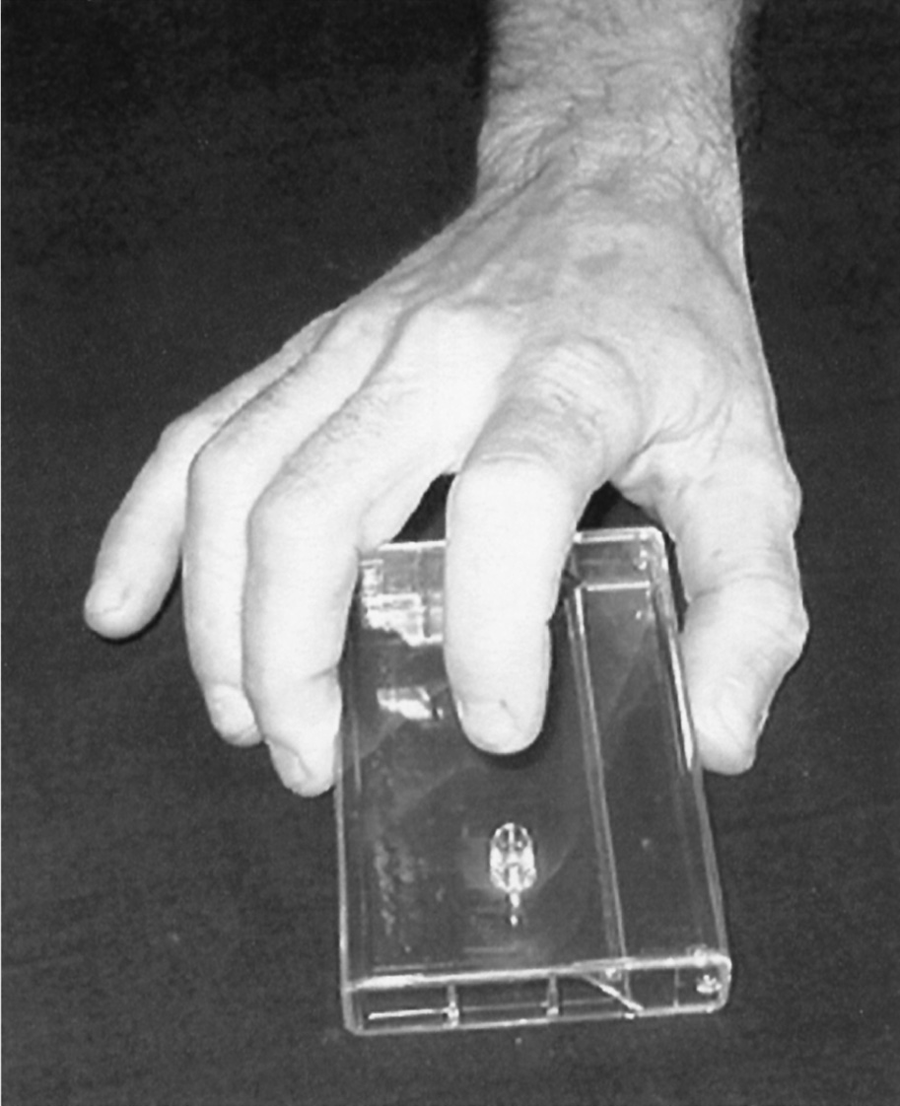
FIGURE 7-42. Position of the patient’s hand for tapping on a plastic audiotape box. The examiner listens for the rate and rhythm of the taps.
b. The deck of cards (or cassette in this example) serves three purposes:
i. The Ex will quickly learn about the Pt’s fine motor skills.
ii. The dyspraxic Pt has difficulty assuming the position or even in picking up and arranging the box.
iii. Most important, the cassette amplifies the sound so that the Ex can estimate the speed and the rhythm. The ear can detect subtle differences between the right and left hands far better than the eye.
c. Because the test engages the entire central and peripheral motor systems, spasticity, rigidity, ataxia, and neuromuscular disorders will slow finger-tapping speed discernibly, and the Ex will easily hear the disturbance in rhythm of the cerebellar Pt.
3. Archimedes spiral
a. Technique: The Pt places a pen in the middle of a sheet of paper and makes a spiral line encircling the center point, making several winds of the pen, out to the periphery.
b. The difference in rigidity that narrows the spiral but leaves its lines fairly regular, ataxia in which the spiral’s lines irregularly weave in and out, and the continuous wavering of lines of essential tremor are striking.
4. Tasks of daily living: Frequently the Ex should elect to watch the Pt button, use scissors, tie shoelaces, drink a glass of water, and arrange clothes as if to put them on.
BIBLIOGRAPHY · Hyperkinesias and Finger Tapping
Arias P, Robles-García V, Espinosa N, et al. Validity of the finger tapping test in Parkinson’s disease, elderly and young healthy subjects: is there a role for central fatigue? Clin Neurophys. 2012;123:2034–2041.
Hallet M, Obeso J. Where does chorea come from? Cortical excitability findings challenge classic pathophysiological concepts. Movt Disord. 2015;30:169–170.
Hawley JS, Weiner WJ. Hemiballismus: Current concepts and review. Parkinsonism Rel Disord. 2012;18:125–129.
Herndon RM, ed. Handbook of Neurologic Rating Scales. New York, NY: Demos Vermande; 2006.
Klein C. Genetics in dystonia. Parkinsonism Rel Disord. 2014;20(S1):S137–S142.
Martino D, Mink JW. Tic Disorders. Continuum (Minneap Minn). 2013;19:1287–1311.
Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. 2015;33:153–174.
Moritz CH, Haughton VM, Cordes D, et al. Whole-brain functional MR imaging activation from a finger-tapping task examined with independent component analysis. Am J Neuroradiol. 2000;21:1629–1635.
Obeso JA, Rodriguez-Oroz MC, Stamelou M, et al. The expanding universe of disorders of the basal ganglia. Lancet. 2014;384:523–531.
Shprecher DR, Schrock L, Himle M. Neurobehavioral aspects, pathophysiology, and management of Tourette syndrome. Curr Opin Neurol. 2014;27:484–492.
Stoessl AJ, Lehericy S, Strafella AP. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet. 2014;384:532–544.
Strauss E, Sherman, EMS, Spreen, O. A Compendium of Neuropsychological Tests: Administration, Norms, and Commentary. 3rd ed. Oxford: Oxford Univ. Press; 2006.
P. Akathisia, restless legs, and hyperactivity: the urge to move
1. Akathisia refers to motor unrest manifested by continual shifting of positions and sometimes by restlessly moving about. When questioned, Pts often report an actual feeling in their muscles of an urge to move (Burkhard, 2014). Akathisia appears typically in Parkinson disease, in other diseases of the basal motor nuclei, and with psychotropic medications (Patel et al, 2014).
2. Restless-legs syndrome (Ekbom syndrome): When attempting to rest or sleep, these Pts feel an irresistible urge, a necessity, to move their legs around. No force of will can hold the legs still against the pathophysiologic imperative, and the aimless, incessant wandering of the legs prevents the onset of sleep. Symptoms are partially or totally relieved by movements such as walking or stretching (Silber et al, 2013). Exhaustive exercise or some drugs, such as antihistamines, may induce restless legs.
3. Attention Deficit/Hyperactivity Disorder (ADHD): It is first diagnosed in children and adolescents by their behavioral symptoms that are severe or out of proportion to their developmental age and without another explanation. Characterized by inattention, hyperactivity and impulsivity, or a combination of these symptoms, it impacts their everyday functions or activities. Symptoms often persist into adulthood and associated with economic and school burdens (eg, failure to complete postsecondary education) (Feldman et al, 2014). In the U.S., it is now the most prevalent neurodevelopmental disorder diagnosed in children (9.5%), but there is also the suggestion that it may be over diagnosed (Coon et al, 2014).
In normal behavioral development executive function allows for the organization of behavior, goal setting and maintenance over time that is reflected in behavior that is goal-directed, self-organized, and flexible. While emotional regulation provides the proper response to motivation rewards as demonstrated by an appropriate emotional response. In ADHD, both of these cognitive skills may be impaired and resulting in the observed behaviors.
There is evidence of a genetic aspect of ADHD that is manifested as an increased incidence among twins and higher risk with another involved family member. Yet, it is less likely that ADHD represents one disorder and more probably a heterogenous condition. Diagnosis continues to rely on symptoms that likely reflect an underlying neurobiological origin; inattention-disorganization predicts academic problems, hyperactivity–impulsivity tend to predict poor social interactions. Functional neuroimaging studies show an association with a delay in cortical maturation, large-scale neural networks and their connectivity (Matthews et al, 2014).
4. Self-mutilation: The Pt compulsively inflicts self-injury by biting, scratching, or pounding despite punishments or rewards. The head-banging infant or child particularly distresses parents. Although generally seen in mentally retarded Pts, some individuals with normal intelligence scratch, bite fingernails, pick their nose or lips, or otherwise injure themselves compulsively (neurodermatitis) in response to some pathophysiologic imperative that overcomes willpower.
5. Stereotyped behavioral mannerisms: Many individuals with intellectual developmental disorder, autistic spectrum disorder, and some otherwise normal children and adults with psychotic disorders display repetitive behaviors, including spinning, rocking, patting, touching, grimacing, licking, and mouthing. Children with Rett syndrome display a characteristic hand wringing. Where these behaviors, in addition to so many of the behaviors discussed above, fall in the scale between voluntary and involuntary further stretches the notion of free will.
Q. Epilepsy
1. Definition: Epilepsy is any change in the mental, motor, or sensory state of the Pt caused by an abnormal hypersynchronous discharge of neurons.
2. Involuntary epileptic motor activity may consist of tonic or clonic spasms and myoclonic jerks that affect all or part of the body or of complex automatisms with laughter and cursive states during complex partial (psychomotor) seizures.
3. If epilepsy causes the abnormal motor activity, the Pt usually will lose consciousness and have amnesia for the episode, and EEG monitoring usually will record epileptiform activity. However, during some epileptiform movements, the Pt retains consciousness (Chapter 13).
R. Avoiding pitfalls (pratfalls) in distinguishing psychogenic from other motility disturbances
1. Psychogenic movement disorders may mimic movement disorders. See Chapter 14.
2. Anxiety or emotional tension makes virtually all hyperkinesias worse, but most dampen or disappear when the Pt is relaxed or asleep. Only a few abnormal involuntary movements occur during sleep: sleep myoclonus, palatal tremor, somnambulism, and some epileptic seizures.
3. During the evolution of one illness, the form of the hyperkinesias or the degree of hypo- or hypertonia may change. Hence, what starts as a flaccid hemiparesis may end as spastic hemiparesis. Simple athetosis may end as dystonia, in which the Pt’s efforts to make a voluntary movement is paralleled by the degree of an athetoid or dystonic spasm. Torsion dystonia is a classic example of a pathophysiologic imperative that determine the contraction of the muscles, rather than the Pt’s “will.”
4. Psychotropic medications, alcohol, and street drugs frequently cause hypokinetic or hyperkinetic movement disorders. Consider drugs in the differential diagnosis of tremors or any other movement disorder of new onset.
5. Parents of children with ADHD invariably appear distraught, depressed, and defeated by their inability to cope with their children. Usually the child’s hyperactivity discloses itself by the slapdash, frenetic way the child executes the Ex’s requests. If, however, the child appears calm during the period of examination, as a few will, the Ex may mistakenly conclude that the parent is “over-anxious.” The usual problem is an “under-anxious” physician, not an overanxious parent. Thus the immediate inspection of the Pt, which serves so well to recognize the standard hyperkinesias of extrapyramidal origin, may, on some occasions, fail in recognizing the hyperactive child if the observation period is too brief. On the next visit, detain the child in the waiting room for a considerable period before the appointment. Then, after the child has defaced your office décor, scattered the toys all over the reception room (without actually playing with any), exasperated your other Pts, and driven your receptionist mad, you will understand the parent’s plight.
S. Patient analysis
The Pt is a mentally normal 47-year-old woman who was born with mild spastic cerebral palsy. Her motor disorder has not changed in the decades since childhood. For the serial photographs, she was requested to extend her arms out in front of her and to hold them as still as possible. Similar movements appear when she is at rest or walking. The individual movements last 2 to 3 seconds and have low to moderate force and amplitude, but may merge with each other in a continuous pattern. The movements shown in Fig. 7-43 would best be classified as _________
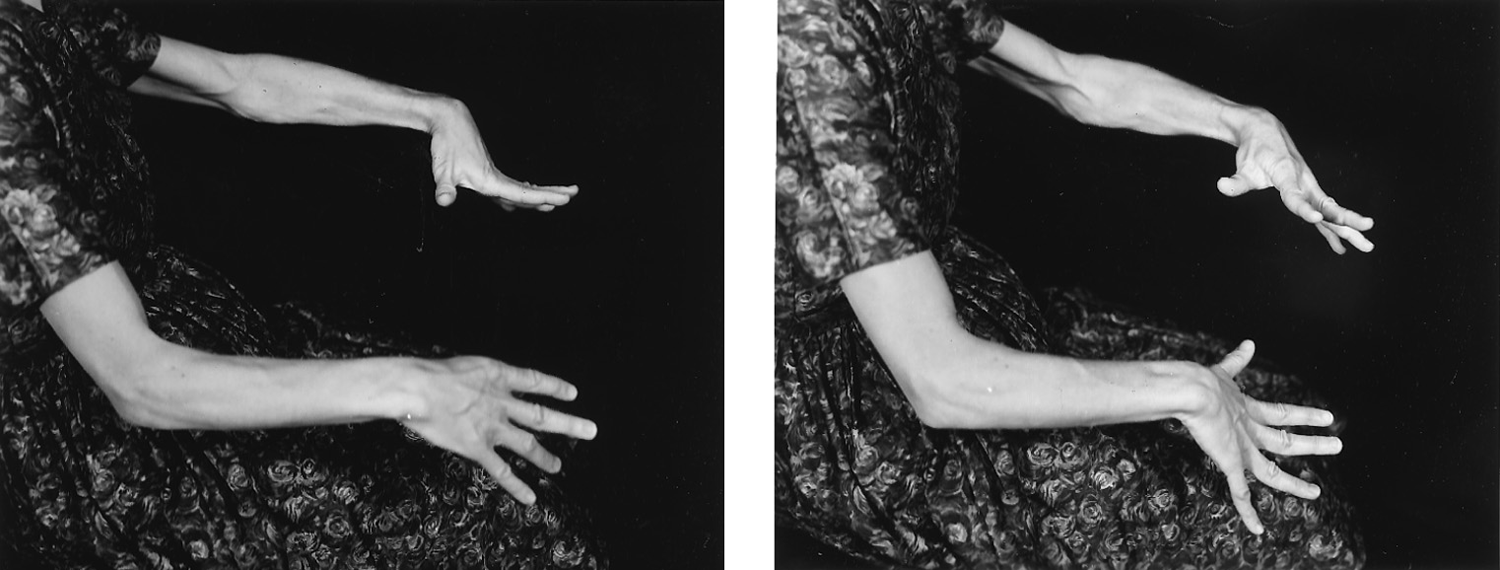
FIGURE 7-43. Action sequence of involuntary movements.
T. A summary of involuntary movements (hyperkinesias) by operational definition
1. For clinical diagnosis, I have defined hyperkinesias broadly to include any extra muscular activity of peripheral or central origin caused by a lesion of the nervous system.
2. For the clinical characteristics in frames a to u, write down the proper descriptive diagnosis in the blank. Where possible, act out the motility disorder described.
a. Spontaneous random contractions of denervated muscle fibers detected by EMG: _________
b. Spontaneous random twitches of small parts of muscles detected by clinical inspection and EMG: _________
c. Sudden spontaneous contraction of a muscle or group of muscles, which may simulate a startle reaction: _________
d. Spontaneous stereotyped sequence of muscular contractions, most prominent in facial muscles, in patients with obsessive-compulsive personality traits: _________
e. Spontaneous tonic or clonic jerking of the body, often accompanied by loss of consciousness: _________
f. Spontaneous, quick movements simulating fragments of normal movements, usually most prominent in extremities: _________
g. Spontaneous, writhing movements of fingers and extremities that may affect facial and axial muscles: _________
h. Spontaneous, long-sustained deviations of appendicular and axial parts, with alternating agonist–antagonist contractions, that may ultimately lead to fixed deformities: _________
i. More or less incessant (during waking hours), wild, flinging movements of one-half of the body, seen usually in elderly hypertensive patients: _________
j. Spontaneous upward deviation of the eyes and head, seen usually with rest tremor and lead-pipe rigidity: _________
k. Irresistible wandering of the legs, especially when the patient tries to rest or to sleep: _________
l. Incessant, driven, usually annoying or aggressive behavior in a child: _________
m. A tremor at rest of 6 cps, which dampens or disappears on intentional movement: _________
n. Irregular tremor of a movement in progress, but no tremor at rest: _________
o. Rapid tremor of an outstretched hand that dampens when a movement is in progress and reappears when a new posture is held: _________
p. A 10-cps tremor of the hands that often has a familial pattern: _________
q. A therapy-resistant hyperkinesia usually with predominant face, lip, and tongue movements that appear after prolonged ingestion of psychotropic medications: _________
r. A state of motor unrest in a Pt with Parkinson disease characterized by irresistible restless shifting of postures: _________
s. Sudden yielding of a sustained posture, as of the dorsiflexed wrist with the arms extended: _________
t. Prolonged rippling or undulating, worm-like contractions of muscles, distributed focally or in widespread groups, associated with repetitive discharges of groups of motor units: _________
u. Irregular, quick, low amplitude, widespread movements of trunk and extremities, often associated with opsoclonus: _________
BIBLIOGRAPHY · Attention Deficit–Hyperactivity Disorder and Involuntary Movements
Burkhard PR. Acute and subacute drug-induced movement disorders. Parkinsonism Rel Disord. 2014;20(S1):S108–S112.
Coon ER, Quinonez RA, Moyer VA, Schroeder AR. Overdiagnosis: How our compulsion for diagnosis may be harming children. Pediatrics. 2014;134:1013–1023.
Feldman HM, Reiff MI. Attention deficit–hyperactivity disorder in children and adolescents. N Engl J Med. 2014;370:838–846.
Matthews M, Nigg JT, Fair DA. Attention deficit hyperactivity disorder. Curr Top Behav Neurosci. 2014;16:235–266.
Patel N, Jankovic J, Hallett M. Sensory aspects of movement disorders. Lancet Neurol. 2014;13:100–112.
Silber MH, Becker PM, Earley C, et al. Willis-Ekbom Disease Foundation Revised Consensus Statement on the Management of Restless Legs Syndrome. Mayo Clin Proc. 2013;88:977–986.
XIII. SUMMARY OF THE SOMATIC MOTOR SYSTEM EXAMINATION
Demonstrate the motor examination, commencing with the initial inspection for gait, posture, tremors, and abnormal movements; followed by palpation, strength testing, and muscle tone; and proceeding through the deep reflexes, percussion, and superficial reflexes.
 Learning Objectives for Chapter 7
Learning Objectives for Chapter 7