12 |
Examination of the Patient Who Has a Disorder of Consciousness |
I may speak alike to you and my own conscious heart.
I. EVALUATION OF CONSCIOUSNESS
A. Two definitions of consciousness: intuitive and operational
1. Intuitively, we define consciousness as the awareness of self and environment. Or, more introspectively, we define consciousness as the awareness of our sensorium.
2. Operationally, physicians determine consciousness by practical steps.
B. Operations that establish consciousness
Physicians customarily employ inspection, verbal stimuli, and, if necessary, pain to determine the patient’s (Pt’s) awareness of self and environment.
1. Inspection: Does the Pt appear to adapt appropriately to the ongoing visual, auditory, and tactile stimuli of the ordinary environment?
2. Verbal stimuli: Does the Pt respond appropriately to inquiries and requests?
3. Pain: Does the Pt respond appropriately to pain?
4. For proof of conscious awareness, the Pt must have a receptor and its sensory pathway intact enough to receive stimuli and deliver those stimuli to the cerebrum. Then the Pt must have a motor pathway, neuromuscular junction, and an effector intact enough to produce a volitional behavior, verbal or nonverbal, that depends on consciousness. The examiner (Ex) cannot determine consciousness by clinical tests in a curarized Pt on the intensive care unit, a Pt with severe Guillain–Barré syndrome (GBS), or a Pt with bilateral hemiplegia, who lacks an intact effector system. See, however, the locked-in syndrome, pages 500–501.
C. Pathologic alterations in the level of consciousness
1. Disease may alter consciousness by causing various stages of delirium or coma.
2. Delirium (acute confusional state): Delirium means an acute, transient confusional state characterized by global impairment of the sensorium. The Pt shows disorientation, amnesia, misperceptions, hallucinations, (often vivid) delusions, brief attention span, disconnected thoughts, irrational or incoherent mutterings, and abnormally decreased or increased psychomotor activity (agitation). Rating scales enable a quantitative assessment (Trzepacz et al, 2002). The sleep and wake periods fail. Any such state of excessive neuronal activity may eventually cause tremors and convulsions (Diagnostic and Statistical Manual of Mental Disorders, 4th ed., 1994; Lipowski, 1989; Victor and Ropper, 2001). The delirious Pts then return to their previous mental state, or delirium may precede coma. Demented Pts may have periods of superimposed delirium. In aged and demented persons, delirium occurs most commonly at night. The causes of delirium include intracranial hemorrhage, infection, sleep deprivation, various drugs or withdrawal from drugs, toxic or metabolic states, and fever. Withdrawal from alcohol or other depressant drugs causes withdrawal delirium (delirium tremens), the classic example of delirium with excitement and often seizures (Chapter 11).
3. Coma (from the Greek word Koma meaning deep sleep) is sustained pathologic unconsciousness resulting from dysfunction of the ascending reticular activating system (ARAS) in the brainstem tegmentum or both cerebral hemispheres. Comatose patients cannot be aroused and their eyes remain closed (The Multi-Society Task Force on PVS, 1994).
4. Depression of consciousness may evolve through several stages or come on abruptly.
5. Recite the minimum neuroanatomic connections necessary for a valid bedside appraisal of consciousness.
_________
_________
D. The Glasgow Coma Scale (GCS) for tracking the level of depressed consciousness
1. A grading scale forces the Ex to observe systematically and accurately. The necessity to grade a response, as in eliciting muscle stretch reflexes (MSRs), makes you observe better than if you do not face such a reckoning.
Depend on it, sir, when a man knows he is to be hanged in a fortnight, it concentrates his mind wonderfully.
2. For the Glasgow Coma Scale (total score 3-15), the Ex periodically grades eye opening from 1 to 4, the best verbal response from 1 to 5, and the best motor response to pain from 1 to 6 (Teasdale and Jennett, 1974). The trend lines derived from these simple tests readily disclose deterioration or improvement. In Fig. 12-1, the trend lines document a declining level of consciousness in a Pt with cerebral edema. After the Pt receives intravenous mannitol, a hyperosmolar agent that reduces edema, the trend lines document the abrupt improvement.
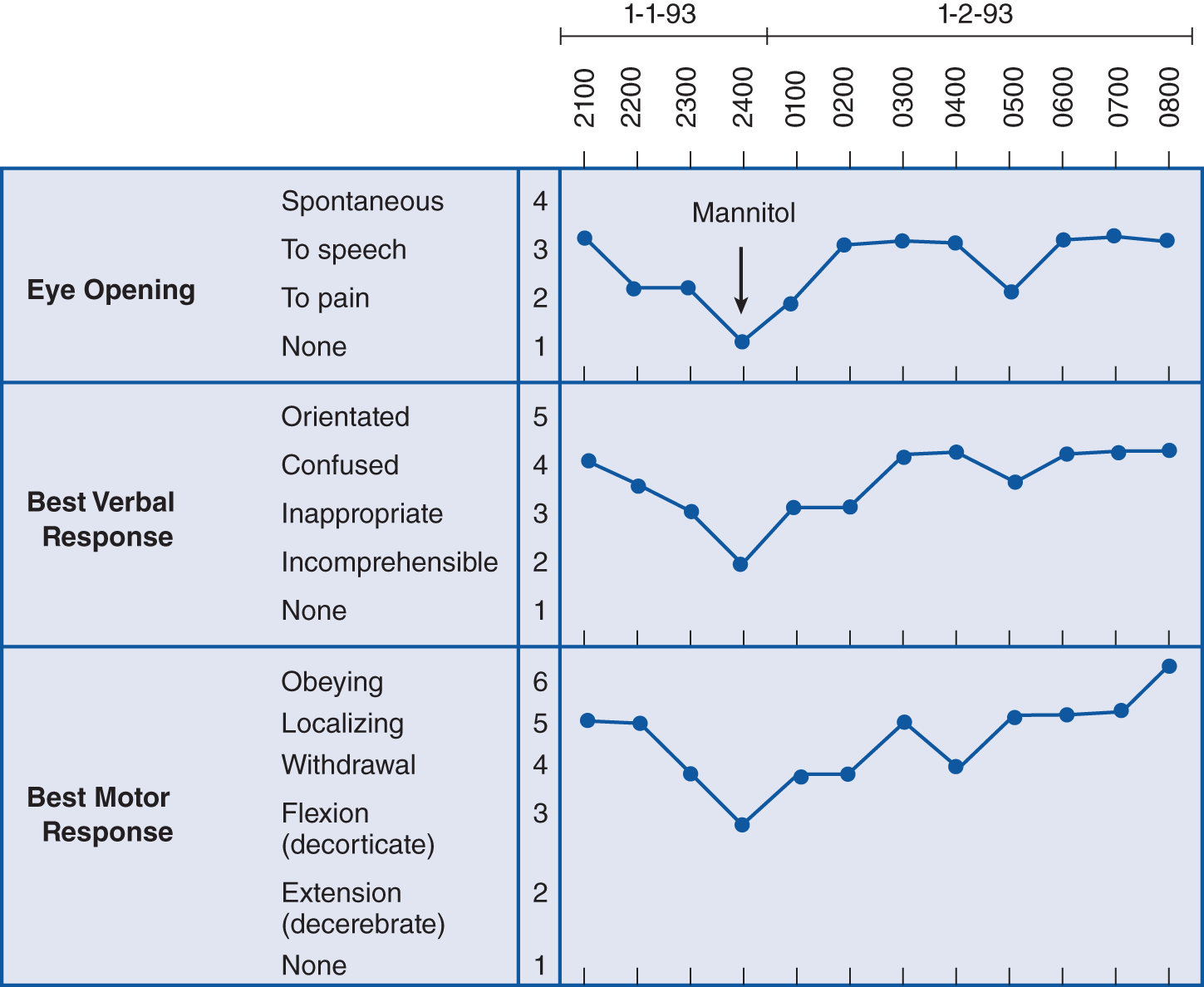
FIGURE 12-1. The Glasgow Coma Scale (GCS). Notice the declining level of consciousness until after mannitol treatment, which reduces brain edema.
3. Like the Apgar score, the total score for the three categories provides a prognosis (Bates, 1991; Evans, 1976; Teasdale et al, 1978). A GCS score from 3 to 8 implies a poor prognosis (Levin et al, 1991). The GCS should report the total score as well as the individual scores for each of the three items of the scale. Valuable as these scales are, some interexaminer variability occurs (Newton et al, 1995; Wiese, 2003).
4. In some intensive care unit Pts with the eyes swollen shut and who are sedated and intubated, the GCS is difficult to apply. Besides, the GCS is insensitive to brainstem function (lack of assessment of pupillary size and reactivity). Thus, modifications of the GCS and new proposals for more reliable coma scales have since been proposed but not universally accepted. The Full Outline of UnResponsiveness (FOUR) score scale (Table 12-1; Wijdicks et al, 2005), proposed as an alternative to the GCS’s, a new scale for improved assessment of coma. The scale has been validated in different settings (Iyer et al, 2009; Stead et al, 2008), and it consists of four components (Eye Response, Motor Response, Brainstem Reflexes, and Respiratory Pattern). The total score is 0 to 16, and each component has a maximal score of 4.
TABLE 12-1 • Full Outline of UnResponsiveness (FOUR) Scale
Eye Response 4 = eyelids open or opened, tracking, or blinking to command 3 = eyelids open but not tracking 2 = eyelids closed but open to loud voice 1 = eyelids closed but open to pain 0 = eyelids remain closed with pain Motor Response 4 = thumbs-up, fist, or peace sign 3 = localizing to pain 2 = flexion response to pain 1 = extension response to pain 0 = no response to pain or generalized myoclonus status Brainstem Reflexes 4 = pupil and corneal reflexes present 3 = one pupil wide and fixed 2 = pupil or corneal reflexes absent 1 = pupil and corneal reflexes absent 0 = absent pupil, corneal, and cough reflex Respiration 4 = not intubated, regular breathing pattern 3 = not intubated, Cheyne–Stokes breathing pattern 2 = not intubated, irregular breathing 1 = breathes above ventilator rate 0 = breathes at ventilator rate or apnea |
SOURCE: Wijdicks et al., 2005. |
II. ANATOMIC BASIS OF CONSCIOUSNESS
A. Parts of the neuraxis unnecessary for consciousness
1. Operationally, the question of the location of consciousness boils down to whether we can locate neuronal circuitry that must be active for consciousness and that, if destroyed, impairs or abolishes consciousness. To identify the parts of the CNS necessary for consciousness, we will first cut away and discard the parts unnecessary for consciousness. To ensure that you know the gross parts of the CNS, make an enlarged drawing of Fig. 2-1 on a loose sheet of paper. Then use scissors to cut away the parts of your drawing, as described next in the text, and place these parts in a pile.
2. First excise the entire spinal cord. Next excise the cerebellum. Then excise the medulla and caudal half of the pons. None of these parts is necessary for consciousness. (Actually cut these parts off of your drawing. This seeming busy work will lead to permanent retention.) Complete transection of the neuraxis at any level from the sacral tip of the cord to the midpons spares consciousness, if we artificially maintain respiration and blood pressure. The pile of parts discarded as unnecessary for consciousness now contains the entire spinal cord, medulla, caudal half of the pons, and the cerebellum. Next, however, complete transection of the brainstem in the upper pons will temporarily abolish consciousness, whereas complete midbrain transection will permanently abolish consciousness.
3. Next, instead of complete transections of the pons and midbrain, let us make partial transections to determine what must remain to support consciousness. To appreciate the partial cuts, review the cross-sectional anatomy of the brainstem by drawing the generalized cross section of the brainstem (Figs. 2-13 and 2-14). Actually cut the parts off of your drawing as called for in the text.
4. We find that complete transection or even removal of the entire basis of the midbrain or pons bilaterally does not abolish consciousness. If we start with a completely intact nervous system, bilateral destruction of the basis of the pons or midbrain spares vertical eye movements but causes complete paralysis of all other volitional movements (Chapter 5). The Pt retains full sensation and full consciousness but can communicate that consciousness by the only available effector mechanism, the vertical eye movements (see the locked-in syndrome). Now snip off the brainstem basis bilaterally and place it on the discard heap.
5. After transecting the basis, we can insert the knife blade a little deeper to transect the medial and lateral lemnisci. The Pt loses the sensations mediated by these pathways, but consciousness remains. Next transect the tectum. Consciousness remains. Next, core out the cranial nerve motor nuclei. Consciousness remains. Thus, from your cross-sectional drawing of the brainstem, cut away the tectum, lemnisci, and cranial nerve nuclei and add them to the discard heap.
6. Now transection of the tegmentum bilaterally between the midpons and rostral midbrain abruptly abolishes consciousness. For consciousness, the midbrain and rostral half of the pontine tegmentum must remain intact (Reznick, 1983) and in continuity with the cerebrum and the diencephalon. Except for the rostral half of the pontomesencephalic tegmentum, we have discarded all other parts of the neuraxis caudal to the diencephalon.
7. From your original drawing, you now have left in your hand a cerebrum in continuity with the diencephalon and pontomesencephalic tegmentum. Now we will determine the role of the remaining parts of the CNS in consciousness by transecting the diencephalon and basal ganglia at successively more rostral levels. We must insert the knife from the bottom of the cerebrum to transect these gray masses bilaterally without damaging the surrounding white matter or cortex. Bilateral transection of the diencephalon permanently and irreversibly abolishes consciousness. As we extend more rostrally into the basal ganglia, the evidence becomes a little less secure because of the lack of pure lesions in human disease; however, early in the history of surgery on the basal ganglia and diencephalon to treat involuntary movement syndromes, neurosurgeons learned not to make bilateral lesions because of the impairment of mentation, consciousness, and speech. Tentatively, we can state that acute bilateral destruction of the globus pallidus and striatum abolishes consciousness—at least, if the lesion extends, as it usually does, a little into the neighboring diencephalon or septal region, or into the neighboring medial hemispheric wall (Freemon, 1971). Thus, we find that bilateral lesions at any level, from the pontomesencephalic tegmentum up through the diencephalon and basal ganglia to the medial hemispheric wall, abolish consciousness (Figs. 12-2 and 12-3).

FIGURE 12-2. Sagittal section of the brain. The stippling shows the part of the pontomesencephalic tegmentum, diencephalon, and basal forebrain in which bilateral lesions abolish consciousness.
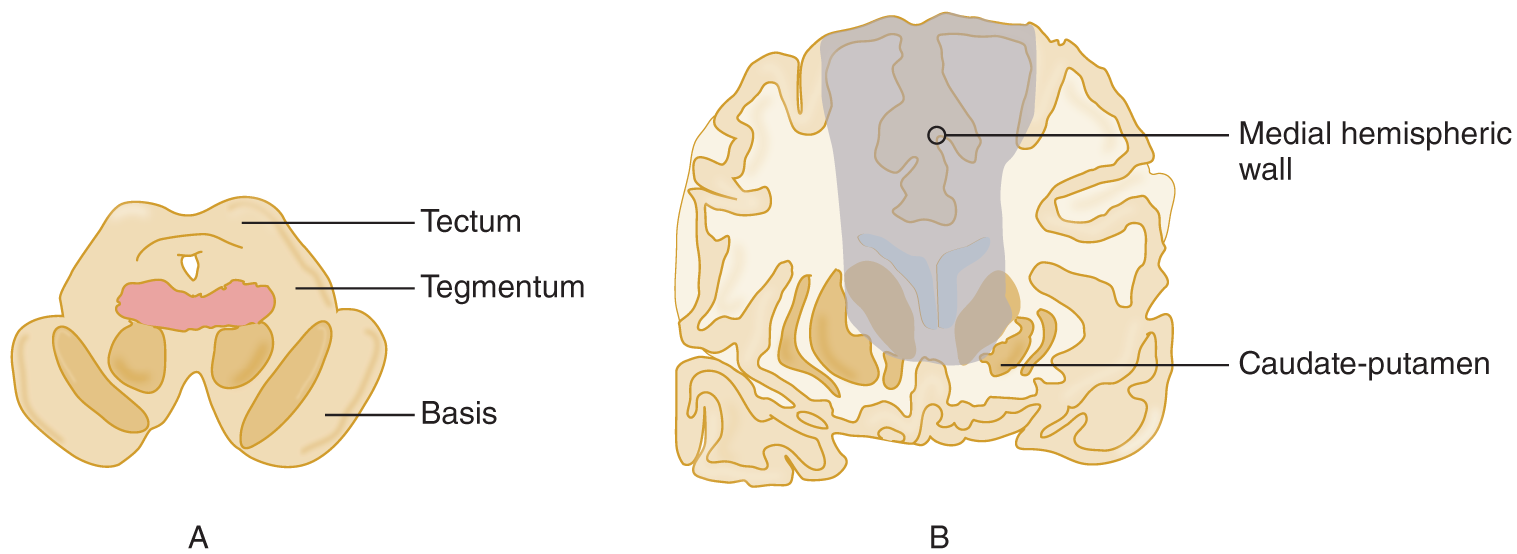
FIGURE 12-3. Coronal sections of the midbrain (A) and the cerebrum (B) at the level of the caudate nuclei. The shaded area shows the sites and comparative sizes of lesions that abolish consciousness.
8. Notice in Figs. 12-2 and 12-3 that, because the lesions involve structures rostral to the midbrain, in particular the basal ganglia and medial hemispheric wall, they must become increasingly larger than those required in the pontomesencephalic tegmentum and diencephalon.
9. The next anatomic region consists of the deep white matter surrounding the diencephalon and basal ganglia, which conveys axonal circuits between those neuronal masses and the cortical neurons. If we destroy the deep white matter of one hemisphere, scrape off all of its cerebral cortex, or remove the white matter and cortex by a hemispherectomy, the Pt can retain consciousness. (However, large acute lesions of a hemisphere temporarily impair consciousness. Curiously, left hemisphere lesions impair consciousness about twice as often as right hemisphere lesions [Albert et al, 1978]). Actually trace a hemisphere from Fig. 12-2, cut it out, and add it to the discard heap. The discard heap of the gross parts unnecessary for consciousness thus includes:
_________
_________
10. Although one of the two cerebral hemispheres is dispensable, we cannot dispense with both. We can discard any pair of the frontal, parietal, occipital, and temporal lobes. We profoundly alter personality and sensorimotor functions, but consciousness per se remains. But if we remove too much of the cerebrum bilaterally, scrape the cerebral cortex off both hemispheres, or destroy much or all of the cortex by hypoxia or hypoglycemia, we produce an apallic syndrome, an old term for a condition that is now considered equivalent to a persistent vegetative state (Dalle et al, 1977). If we suck out the deep white matter from both hemispheres, or if the Pt has a severe demyelinating disease, thus disconnecting the cortical shell from the brainstem and diencephalon, the Pt permanently loses consciousness. Such severe, bilateral destructive decorticating or demyelinating lesions stand in direct contrast to the tiny, confined, bilateral pontomesencephalic tegmental lesions that exquisitely and selectively abolish consciousness, with little effect on functions mediated through other pathways. Review Fig. 12-3, which shows the location in the midbrain tegmentum of the smallest, most discrete lesion that will permanently abolish consciousness.
B. Summary of the parts of the neuraxis necessary and unnecessary for consciousness
1. The previous observations show that full consciousness requires the pontomesencephalic tegmentum of at least one-half of the brainstem and the ipsilateral diencephalon and some of the ipsilateral cerebral hemisphere. This may, just may, constitute the minimal amount of brain required to function as a person. Although a necessary condition for consciousness, an intact pontomesencephalic tegmentum alone is not a sufficient condition or at least not sufficient to provide operational evidence of consciousness. That requires, in addition, a cerebral hemisphere or most of a hemisphere with an output pathway to some effector to produce behavioral responses that signify consciousness. What is not expendable bilaterally without profoundly affecting or abolishing consciousness is
a. Medial hemispheric wall down to and including basal forebrain
b. Caudate–putamen (striatum)
c. Diencephalon
d. Midbrain tegmentum: Bilateral tegmental lesions of small size completely and permanently abolish consciousness
e. Rostral pontine tegmentum
2. To justify your having learned the structures unnecessary for consciousness, note this supreme fact: If your Pt has a lesion confined to one of these structures and is or becomes unconscious, something else has caused the unconsciousness. The usual cause is that the lesion has herniated or shifted the brain to compress the structures indispensable for consciousness. Without medical or surgical intervention, the Pt will die. Section III discusses these brain herniations and their clinical recognition.
C. To prove your knowledge of the neuroanatomic basis of consciousness
1. Enumerate the parts of the neuraxis that we can discard without abolishing consciousness.
_________
_________
2. Enumerate the various sites of lesions within the neuraxis that will abolish consciousness. Assume artificial support of breathing and blood pressure to ensure that the neural lesion causes loss of consciousness, not hypoxia or ischemia.
_________
_________
3. State where to put the smallest lesion that will most selectively abolish consciousness.
_________
_________
D. Operational demonstration of the pathways for consciousness
1. Insert a stimulating electrode into the midbrain reticular formation of an animal.
2. Apply recording electrodes over the scalp. The record obtained from the scalp electrodes is called an electroencephalogram (EEG).
3. After the animal goes to sleep, stimulate the reticular formation and observe:
a. That the animal opens its eyes and looks around.
b. That the EEG shows a distinct change in the electrical activity of the brain (Fig. 12-4).
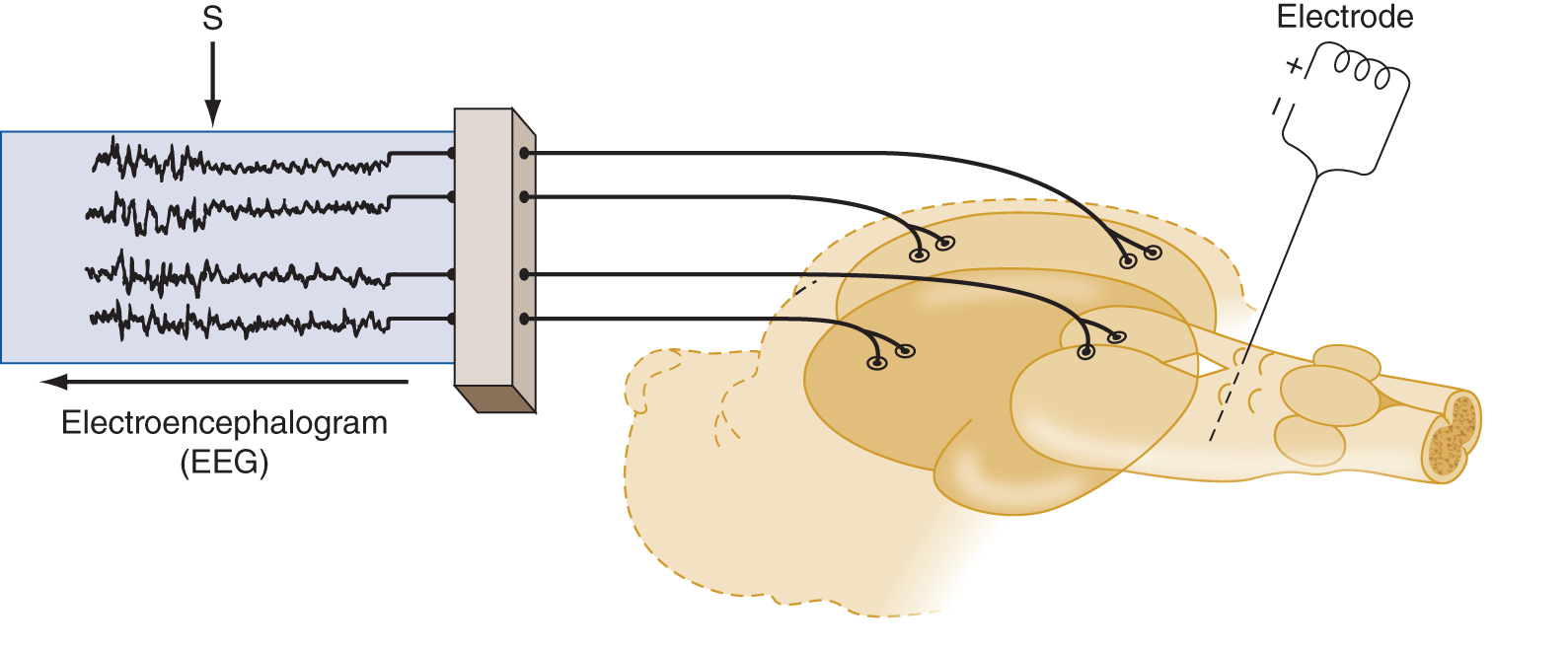
FIGURE 12-4. A sleeping animal with an electrode inserted into the midbrain reticular formation is hooked to an EEG machine. Stimulation of the electrode causes an abrupt awakening. S marks the point of stimulation during the EEG recording. Notice the abrupt change in the entire EEG from the high-amplitude slow waves of sleep to the low-amplitude fast activity of the waking state. EEG = electroencephalography.
4. After having awakened the animal with a stimulus to the reticular formation, the experimenter can greatly increase the current to make an electrolytic lesion around the tip of the electrode. The animal will lapse into unconsciousness.
5. Similar stimulation of the midline and intralaminar nuclei of the thalamus will produce an alerting response. Bilateral destruction abolishes consciousness.
6. Interpretation of the experiment: Demonstration of a nonspecific ascending pathway.
a. The initial stimulation through the electrode was like throwing on a master switch: The entire cortex lit up. Because the entire cortex responded, we say that a diffuse or nonspecific ascending pathway has been stimulated. Because these impulses run through the thalamus, we conclude that the experiment demonstrates an ascending reticulo–thalamocortical pathway. We call this entire pathway the ascending reticular activating system (ARAS). Although the physiologic evidence points to a thalamocortical projection of the ARAS, the actual axons have yet to be fully demonstrated. The axons of some of the chemically specified reticular formation nuclei that belong to the ARAS bypass the thalamic nuclei.
b. Notice that the operational definition of the ARAS depends on two lines of evidence: Stimulation of the system  heightens/ decreases consciousness, whereas destruction heightens/ decreases consciousness. (
heightens/ decreases consciousness, whereas destruction heightens/ decreases consciousness. ( heightens; decreases)
heightens; decreases)
7. Give an operational definition of the ARAS based on the effects of stimulation or destruction._________
_________
E. Demonstration of specific thalamocortical pathways
1. The thalamocortical pathways of the sensory nuclei have a very specific, point-to-point relation to the cerebral cortex, in contrast to the ARAS, which projects diffusely or nonspecifically. To demonstrate this contrasting, specific system, do the following:
a. Insert a stimulating electrode into one of the sensory relay nuclei of the thalamus.
b. Apply recording electrodes over the cortical receptive area of the nucleus. In Fig. 12-5, the stimulating electrode is inserted into the nucleus ventralis posterior, and the recording electrodes are placed over the somesthetic receptive area in the postcentral gyrus. Study Fig. 12-5.
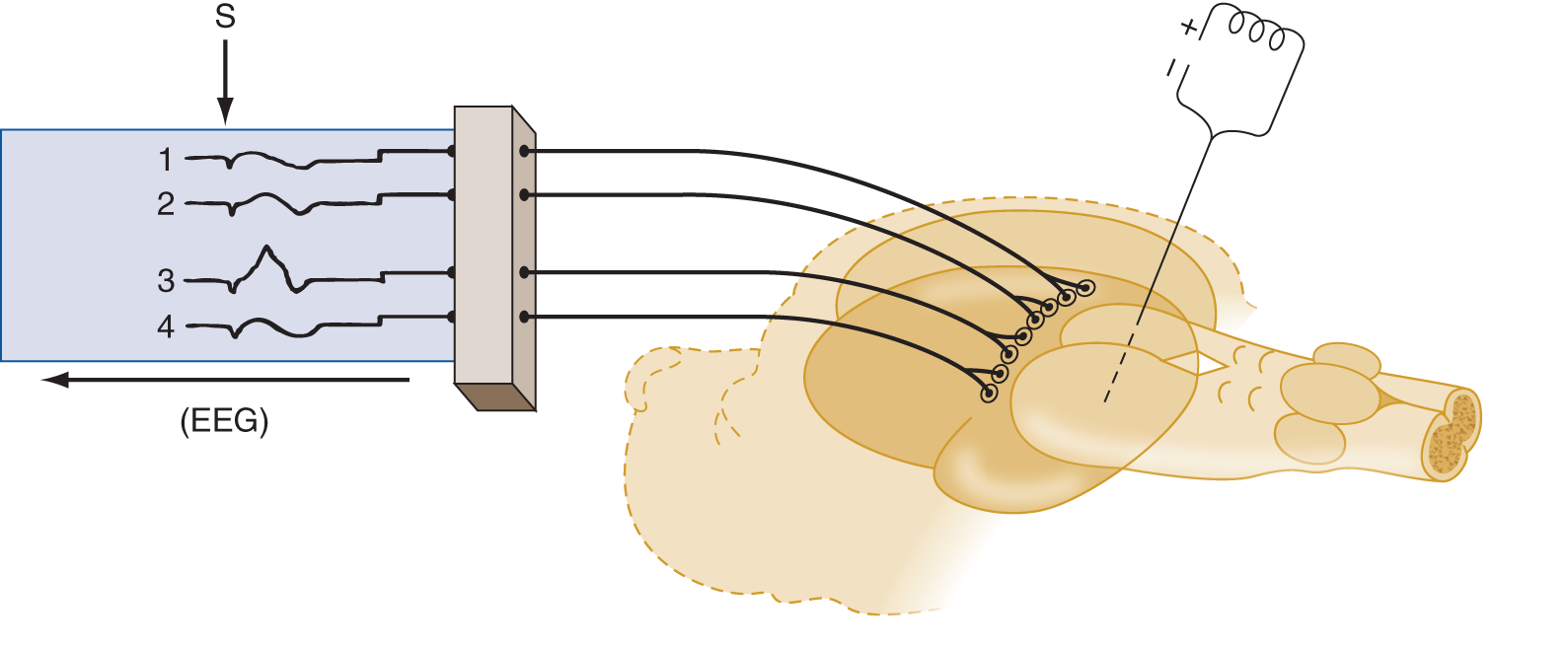
FIGURE 12-5. An electrode stimulated after insertion into one specific site in a thalamic somatosensory relay nucleus causes only one specific point of cortical excitation, in lead 3 of the electroencephalogram, rather than a generalized responses as in Fig. 12-4.
2. The specific systems are so specific that stimulation of minute retinal areas, stimulation of the cochlea by discrete sound frequencies, or stimulation of the skin of the individual digits evokes a response in a tiny, specific cortical sensory area. Review the specific thalamic sensory relay nuclei and their cortical projection areas in Table 12-2.
TABLE 12-2 • Specific Sensory Relay Nuclei of the Thalamus
Modality |
Thalamic nucleus |
Cortical receptive area |
Vision |
Calcarine cortex in _________ |
|
Hearing |
Superior temporal gyrus in _________ |
|
Somatic sensation |
Postcentral gyrus in _________ |
(lateral geniculate body; occipital-medial geniculate body; temporalnucleus ventralis posterior; parietal)
3. The specific/ nonspecific pathways of the ARAS mediate the general state of consciousness, whereas the specific/ nonspecific thalamocortical pathways mediate consciousness of particular sensory events. (nonspecific; specific)
BIBLIOGRAPHY · Consciousness
Albert ML, Silverberg R, Reches A, et al. Cerebral dominance for consciousness. Arch Neurol. 1978;33:453–454.
Bates D. Defining prognosis in medical coma. J Neurol Neurosurg Psychiatry. 1991;54:569–571.
Dalle OG, Gerstenbrand F, Lucking CF, et al. The apallic syndrome. Berlin, Germany: Springer-Verlag; 1977.
Evans BM. Patterns of arousal in comatose patients. J Neurol Neurosurg Psychiatry. 1976;39:392–402.
Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34:693–698.
Iyer VN, Mandrekar JN, Danielson RD, et al. Validity of the FOUR score coma scale in the medical intensive care unit. Mayo Clin Proc. 2009;84(8):694–701.
Levin HS, Williams DH, Eisenberg HM. Serial MRI and neurobehavioral findings after mild to moderate closed head injury. J Neurol Neurosurg Psychiatry. 1991;55:255–262.
Lipowski ZJ. Delirium in the elderly patient. N Engl J Med. 1989;52:578–582.
Newton CRJC, Kirkham FJ, Johnston B. Inter-observer agreement of the assessment of coma scales and brainstem signs in non-traumatic coma. Dev Med Child Neurol. 1995;37:807–813.
Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s Diagnosis of Stupor and Coma. Oxford: Oxford Univ. Press; 2007.
Plum F, Posner J. The Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.
Reznick M. Neuropathology of seven cases of locked-in syndrome. J Med Sci. 1983;60:67–68.
Stead LG, Wijdicks EFM, Bhagra A, et al. Validation of a new coma scale, the FOUR Score, in the Emergency Department. Neurocritical Care. 2008;10(1);50–54.
Teasdale G. Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2:81–84.
Teasdale G, Knill-Jones R, van der Sande J. Observer variability in assessing impaired consciousness and coma. J Neurol Neurosurg Psychiatry. 1978;41:603–610.
The Multi-Society Task Force on PVS. Medical aspects of the persistent vegetative state—first of two parts. N Engl J Med. 1994;330:1499–1508.
Trzepacz PT, Meagher DJ, Wise MG. Neuropsychiatric aspects of delirium. In: Yudofsky SC, Hales RE, eds. Textbook of Neuropsychiatry and Clinical Neuroscience. 4th ed. Washington, DC: American Psychiatric Publishing; 2002, Chap. 14, 525–564.
Victor M, Ropper AM. Adams and Victor’s Principles of Neurology. 7th ed. New York; NY: McGraw-Hill; 2001.
Wiese MF. British Hospitals and different versions of the Glasgow Coma Scale: telephone survey. BMJ. 2003;327(418);782–783.
Wijdicks EFM, Bamlet WR, Maramattom BV, et al. Validation of a new coma scale. The FOUR Score. Ann Neurol. 2005;58:585–593.
III. INTERNAL HERNIATIONS OF THE BRAIN: EFFECT ON CONSCIOUSNESS, NEUROLOGIC FUNCTION, AND VASCULAR SYSTEM
A. Causes and consequences of internal herniations
The most common causes of internal herniations of the brain are cerebral contusions, hematomas, abscesses, neoplasms, and cerebral edema. The mass effect of these lesions increases the intracranial pressure (ICP) and causes internal shifts or herniations of the brain that compress the normal tissue, in particular the diencephalon and brainstem (Cuneo et al, 1979; Davis and Robertson, 1991; Posner et al, 2007; Sunderland, 1958; Walker, 1963). This compression impairs consciousness and the life-sustaining functions of breathing, blood pressure control, and temperature regulation. Herniations also compress cerebral arteries, resulting in infarctions.
B. Anatomy of the intracranial partitions and compartments
To understand how space-occupying lesions act to cause coma or kill Pts, learn Figs. 12-6 to 12-8, and complete the subsequent exercises.
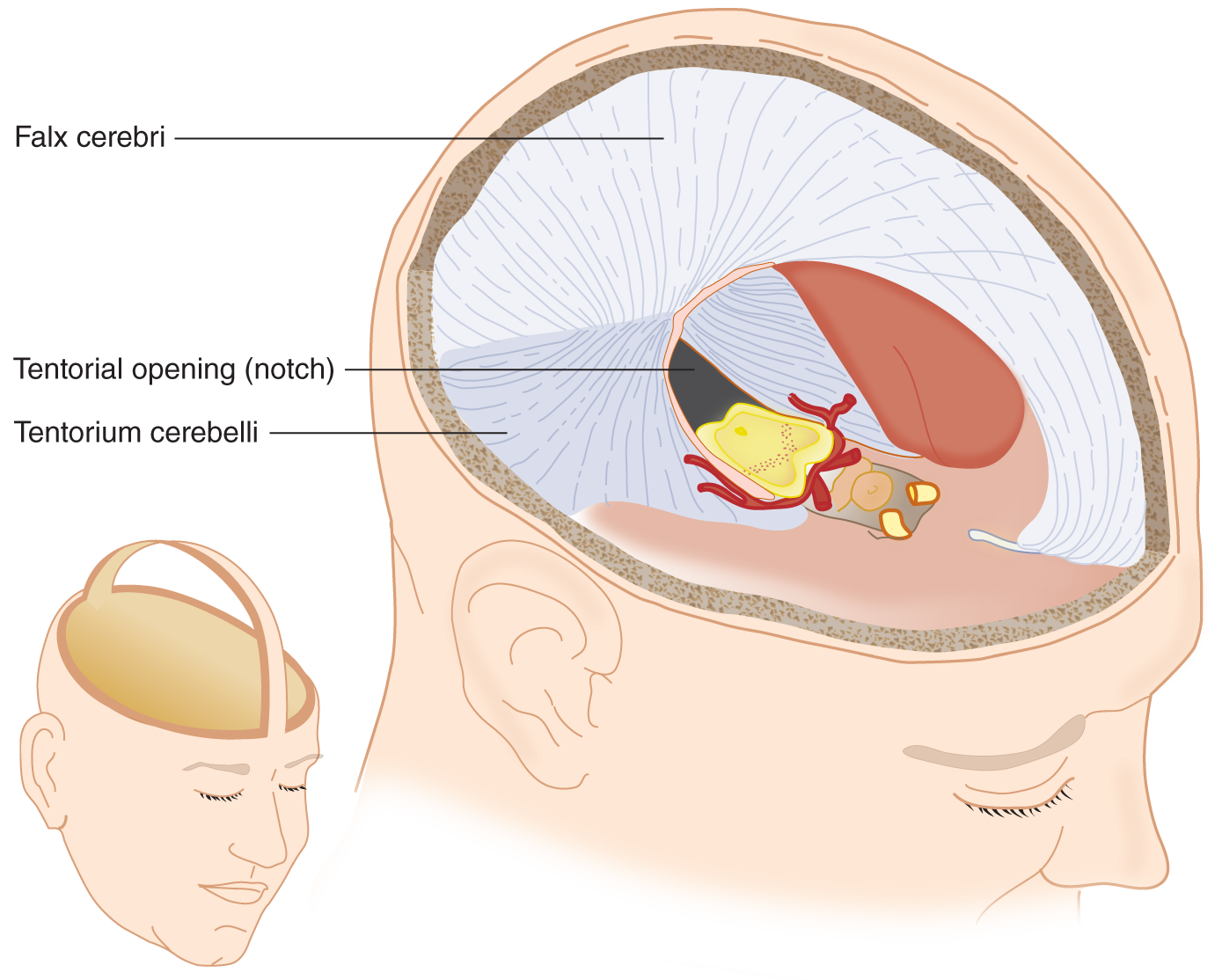
FIGURE 12-6. Basket-handle dissection of the skull and removal of the cerebral hemispheres. Notice that the flax cerebri and tentorium cerebella, folds of the dura mater, partition the intracracterior cerebral arteries course along the ventral edge of the midbrain and extend laterally to cross the free edge of the tentorium to reach the temporo-occipital portions of the cerebrum. Locate the IIIrd nerve issuing from under the posterior cerebral artery. See also Fig. 12-7.
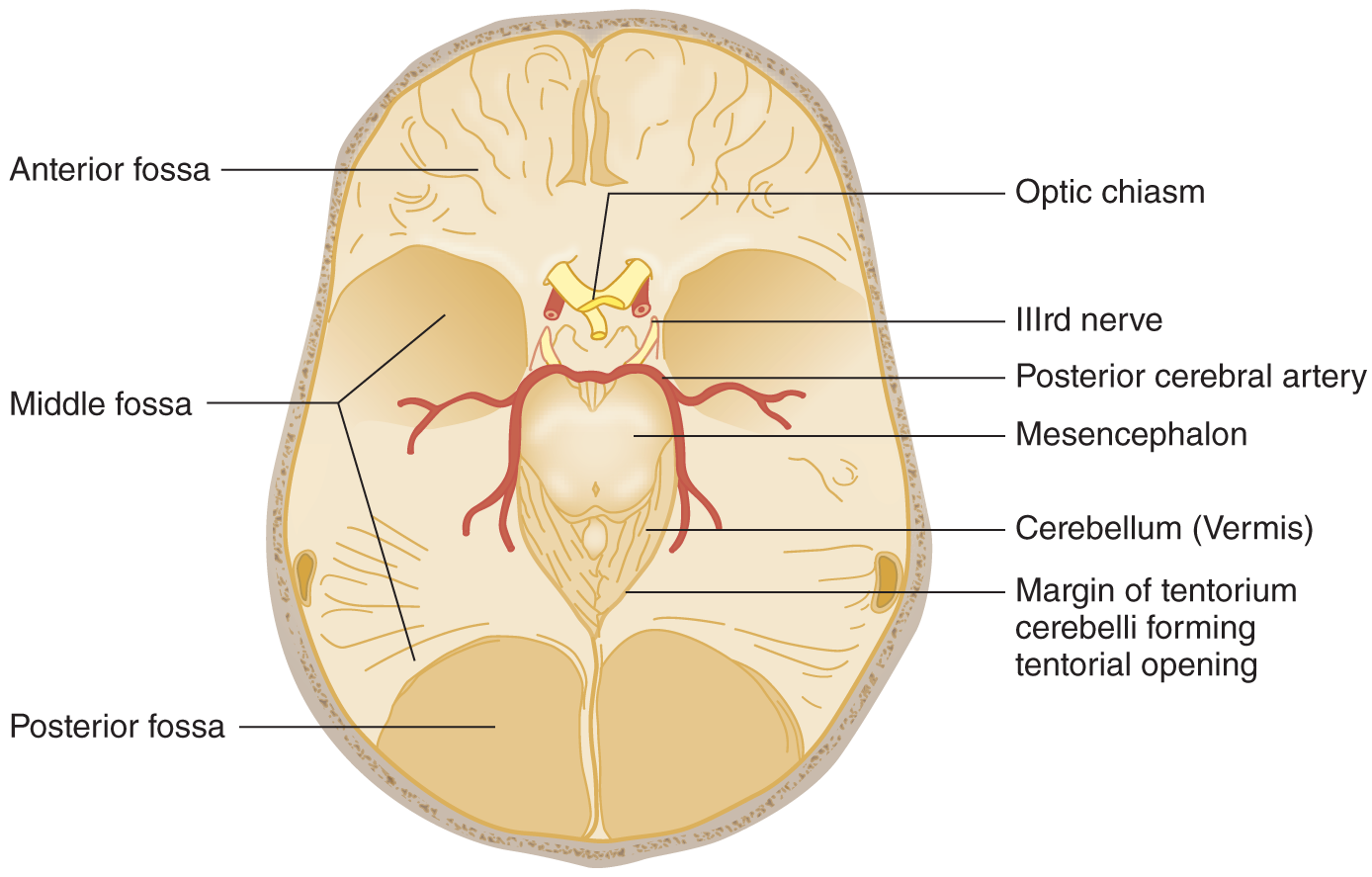
FIGURE 12-7. Base of the skull after removal of the basket handle and its attached falx cerebri. The mesencephalon remains in situ, as in Fig. 12-6. Notice the IIIrd nerve traveling under the PCA = Notice that the cerebellar vermis occupies the apex of the tentorial opening. The posterior fossa is under the tentorium. PCA posterior cerebral artery. (Adapted with permission from Plum F, Posner J. The Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.)
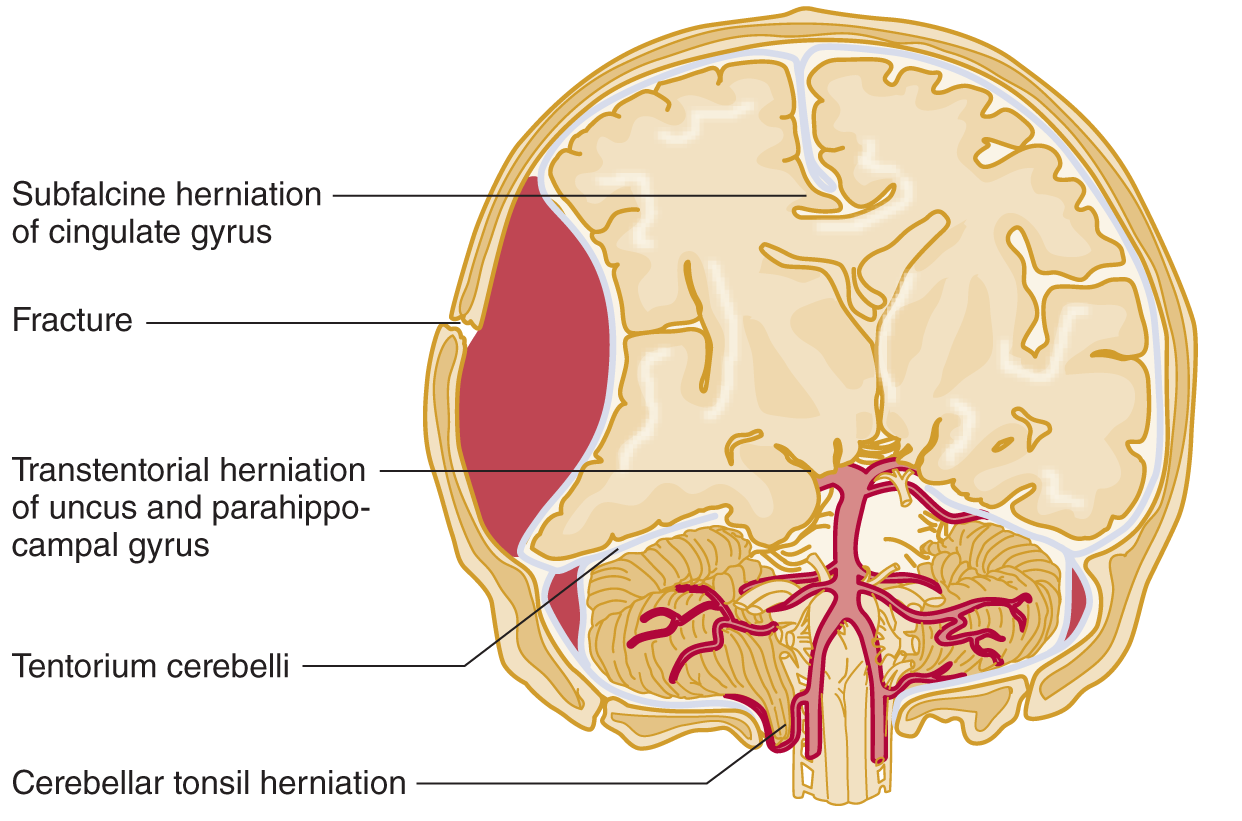
FIGURE 12-8. Coronal section of the head; ventral view of the brainstem and cerebrum. A large epidural hematoma has shifted the brain from right to left. Notice the herniation of the right uncus over the tentorial edge. Follow along the vertebral and basilar arteries to their terminal branches, the posterior cerebral arteries. Notice how the posterior cerebral artery on the right impinges on the IIIrd nerve. (Adapted with permission from Chapter 8. Head and Neck. In: Hansen, J. T. Netter’s Clinical Anatomy 3E. Philadelphia, PA: Saunders/Elsevier, 2014.)
1. The cranial cavity is divided by tough, dural partitions called the _________
2. The dural partition that divides the supratentorial space into right and left halves is the _________
3. The tentorium cerebelli forms a tent over the cerebellar hemispheres by inserting itself between the cerebellar hemispheres below and the _________
4. The space between the free, medial edges of the tentorial halves is called the tentorial _________
5. The part of the cerebellum that protrudes through the tentorial notch is the superior tip of the _________
6. The tentorial notch surrounds the pons/ mesencephalon/ diencephalon. (mesencephalon)
7. In Figs. 12-6 and 12-7 notice that:
a. The transected brainstem remains in situ after removal of the cerebral hemispheres and diencephalon. Notice that the posterior cerebral artery passes over the free edge of the tentorial notch to reach the temporo-occipital portions of the cerebrum.
b. CrN III issues from the mesencephalon and passes under the posterior cerebral artery.
C. Responses of the fluid pools of the intracranial space to increased pressure
1. Water comprises 75% of the brain. Water is physically incompressible and biologically relatively immobile, although in cerebral edema it may increase the brain volume by 20% to 30%. This relatively immobile brain water contrasts with the two rapidly mobile liquid pools of the intracranial space, the intravascular blood and the cerebrospinal fluid (CSF). If brain tissue swells, what will happen to the lumen of the veins and capillaries? _________
2. After studying Figs. 12-8 and 12-9, describe what pressure does to the ventricles, sulci, and subarachnoid spaces of a swollen hemisphere. _________
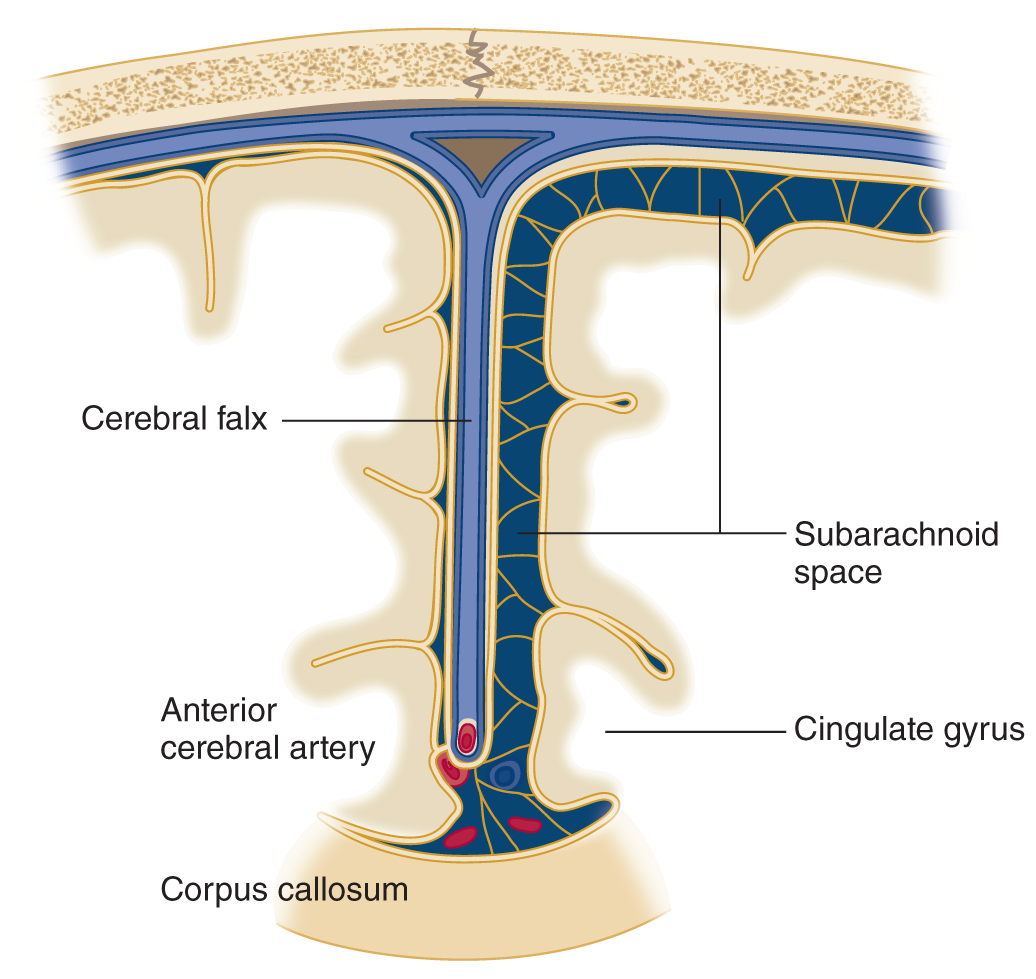
FIGURE 12-9. Coronal section through the falx and adjacent portions of the cerebrum along the interhemispheric fissure. Notice that the swollen hemisphere (reader’s left) has collapsed the subarachnoid space and that the cingulate gyrus has begun to herniate under the free, inferior edge of the falx, impinging on the anterior cerebral artery.
3. Thus, the first compensation for increased pressure is a reduction in the two rapidly mobile intracranial fluid pools, the _________
4. The infant has additional ways to compensate for increased intracranial pressure, manifested by the following physical signs:
_________
_________
5. As the skull matures, it no longer yields to increased intracranial pressure. If you find split sutures on a skull radiograph of an 18-year-old Pt, the increased pressure must have started before the age of _________
6. If the hemispheric swelling exceeds the compensatory mechanisms in an infant or adult, the affected tissue can only respond by herniating. Study Fig. 12-8 and state the only two places the shifting hemisphere can go _________
_________
7. Because part of the swollen hemisphere has shifted under the falx cerebri, this shift is called _________
D. Anatomy of transfalcine herniation
1. The part of the hemisphere that shifts under the falx is called the _________
2. The anterior cerebral arteries run parallel to the free edge of the falx. When the cingulate gyrus herniates, it may compress the artery against the free edge of the falx, causing infarction of the medial hemispheric wall dorsal to the corpus callosum (Fig. 12-9).
3. In view of the representation of body parts in the motor cortex on the medial aspect of a hemisphere (Fig. 2-2C), this infarction would cause upper motoneuron (UMN) paralysis of the leg/ arm/ face/ leg, arm, and face. (leg)
E. Anatomy of transtentorial herniation of the cerebrum
1. With a space-occupying lesion in one hemicranium, the bony wall of the calvarium prevents herniation or decompression outward. The cerebral hemisphere can herniate only medially or downward (Fig. 12-8).
a. The dural membrane that opposes medial shift of the cerebrum is the _________
b. The dural membrane that opposes downward shift of the cerebrum is the _________
2. Trans means over or across, hence, trans continental. The shifting of a swollen hemisphere across the free edge of the tentorium cerebelli is called _________
3. Hence, the two internal herniations of a swollen hemisphere are _________
4. The parts that undergo transtentorial herniation are the medial parts of the temporal lobe, namely the _________
5. Figure 12-10 shows a brain removed at autopsy. Fill in the labels and, in the blanks below, describe what is wrong.
_________
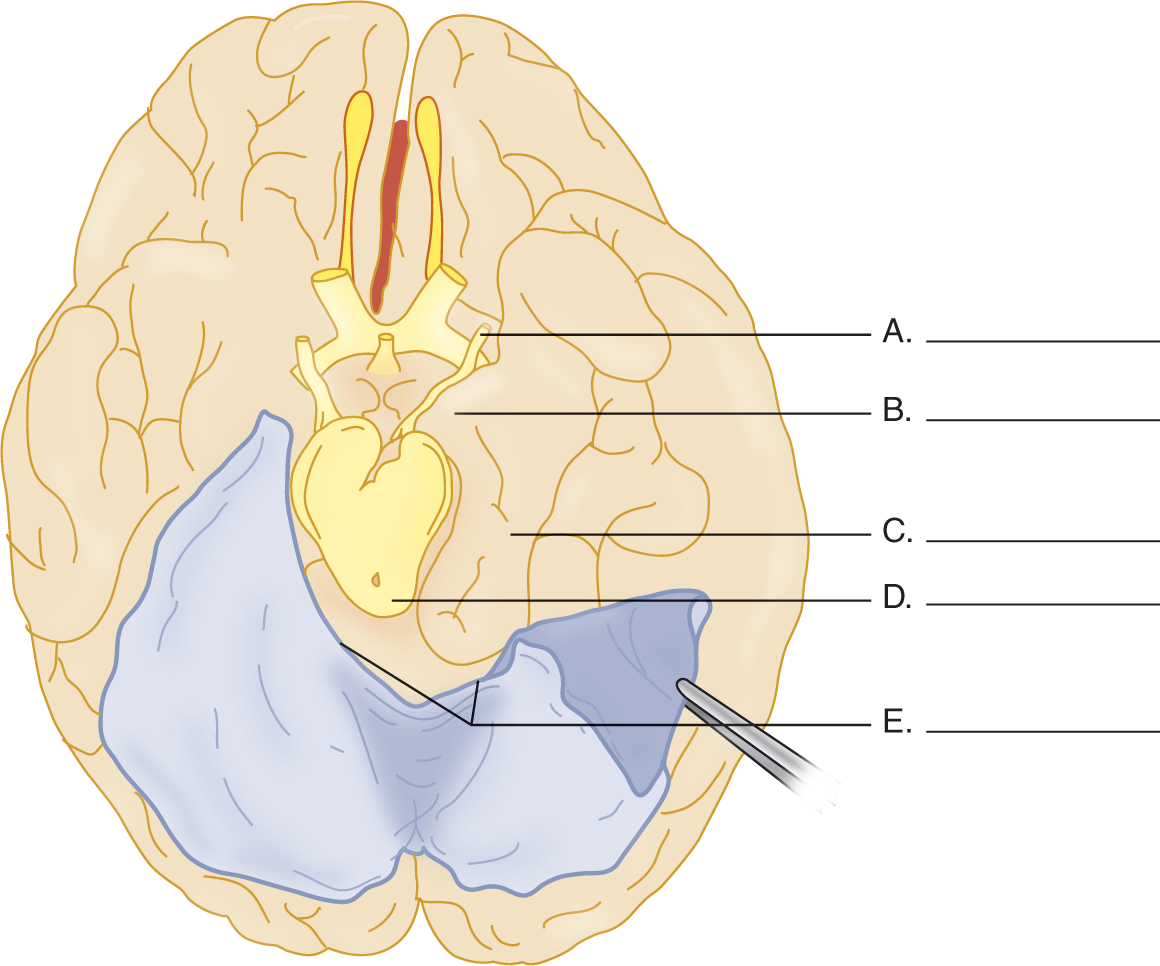
FIGURE 12-10. Ventral view of the cerebrum with the tentorium cerebelli reflected on the left side. Complete the blanks on the right side. (Redrawn from Peele T. The Neuroanatomic Basis for Clinical Neurology, 2nd ed. New York, McGraw-Hill, 1961.)
6. In coursing from the basilar artery to the cerebrum, the posterior cerebral artery crosses the free edge of the falx, where the herniation may compress it (Figs. 12-6 and 12-7 and Table 12-5). This artery irrigates the medial part of the temporo-occipital region and the occipital pole. Infarction of this region, particularly on the right, would cause two ophthalmologic disorders, _________
F. Effect of transtentorial and subfalcine herniation on consciousness
Because subfalcine and transtentorial herniations compress the medial hemispheric wall, diencephalon, and mesencephalon, they interfere with the ARAS and alter the Pt’s level of consciousness. Medial shift of the brain alone may impair consciousness (Fisher, 1995; Inao et al, 2001; Pullicino et al, 1997; Ropper, 1993) but, if combined with transtentorial herniation, compresses the brainstem and may kill the Pt.
G. Effect of transtentorial herniation on cranial nerve III
1. As transtentorial herniation increases, the uncus displaces the posterior cerebral artery and CrN III. CrN III contains parasympathetic fibers that, when stimulated, cause the pupil to _________
2. After compression of CrN III, what happens to the pupillary size? Explain.
_________
_________
3. Because the pupilloconstrictor fibers occupy the superomedial part of CrN III, the caudally displaced posterior cerebral artery impinges on them first when the temporal lobe herniates (Fig. 12-11).
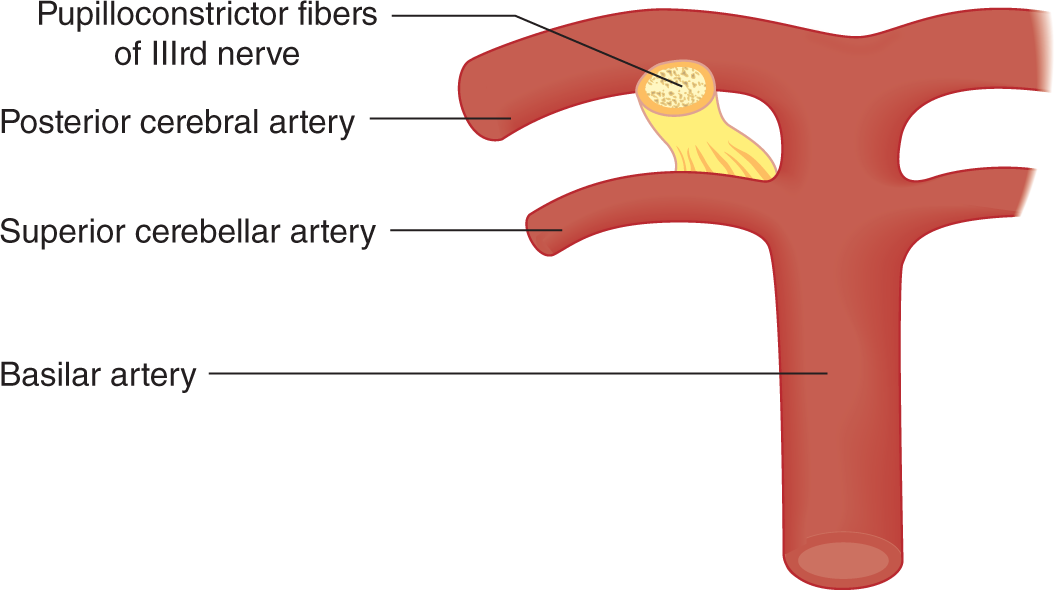
FIGURE 12-11. Relation of pupilloconstrictor fibers of cranial nerve III to the posterior cerebral artery. To orient this illustration, see Figs. 12-6 and 12-8.
4. In a Pt suspected of having a space-occupying intracranial lesion, what would an increasing pupillary size and a decreasing level of consciousness imply?
_________
_________
5. As transtentorial herniation advances, both IIIrd nerves may cease to function. Both pupils become dilated and fixed, no longer responding to light. Before dilating, the opposite pupil may go through a phase of mild constriction (Forbes et al, 1965; Ropper, 1990). Whereas a unilateral dilated and fixed pupil announces danger requiring prompt or even heroic intervention, bilateral dilated and fixed pupils from brainstem compression are almost synonymous with death without prompt neurosurgical intervention (Clusmann et al, 2001).
H. Effect of transtentorial herniation on motor function
1. Consider a Pt with a large acute right hemisphere lesion. It would cause a left-sided/ right-sided hemiplegia (Video 12-1). (left-sided)

Video 12-1. Recovery from right malignant middle cerebral artery (MCA) stroke; status posthemicraniectomy.
2. As the herniating right hemisphere compresses the mesencephalon, the left basis gets pushed against the opposite free edge of the tentorium on the left side, indenting the basis (Kernohan–Woltman notch). Notice in Fig. 12-7 the close relation of the basis mesencephali to the tentorial edges, affording virtually no safety factor.
3. After compression of the left basis, the UMN fibers cannot transmit impulses. The Pt now shows a _________ ipsilateral/ contralateral to the right hemisphere lesion. (right; ipsilateral)
4. Such an ipsilateral hemiplegia is called a paradoxical hemiplegia because it is on the same/ opposite side as the hemispheric lesion. Sometimes the paradoxical hemiplegia appears first, constituting a false localizing sign of the side of brain herniation. (same)
5. Hence, if a Pt started out with a large right hemispheric lesion and left hemiplegia and then had a right hemiplegia, the Pt has double hemiplegia. If the double hemiplegia evolves very rapidly, the Pt might display cerebral shock. What would happen to the MSRs and tone? _________
6. Thus, depending on the rapidity of evolution of the lesion, the MSRs and tone may change from time to time and from side to side.
7. Recite the neurologic findings in transtentorial herniation with respect to the IIIrd nerve, the ARAS, and the pyramidal tracts in the basic mesencephali. Check the preceding frames to be sure that you have recited the effects correctly.
8. The Kernohan-Woltman notch/paradoxical hemiplegia sequence can be regarded as a contrecoup condition because it is on the opposite side of the inciting hemispheric lesion; but in acute head injury a coup type of displacement may similarly nick the midbrain ipsilaterally (Saeki et al, 2000).
I. Bilateral transtentorial herniation
1. Many pathologic conditions cause bilateral transtentorial herniation. The lesion may consist of cerebral edema from trauma, encephalitis, or metabolic disorders such as uremia and hepatic coma. Of the structural lesions, head injuries, subdural hematomas, hydrocephalus, multiple metastatic neoplasms or abscesses, and intracranial hemorrhage lead the list. If both hemispheres swell, the unci and parahippocampal gyri of both sides try to squeeze down through the tentorial notch. The ring of swollen tissue acts exactly like a ligature around the midbrain.
2. The coup de grace to the Pt with transtentorial herniation, bilateral or unilateral, is mesencephalic and pontine hemorrhages secondary to the herniation (Friede and Roessmann, 1966; Hassler, 1967). The stretching and compression of the brainstem vessels stops the blood flow. The vessel walls rupture. The consequent brainstem hemorrhage is usually the terminal event that causes the Pt to die (Fig. 12-12).
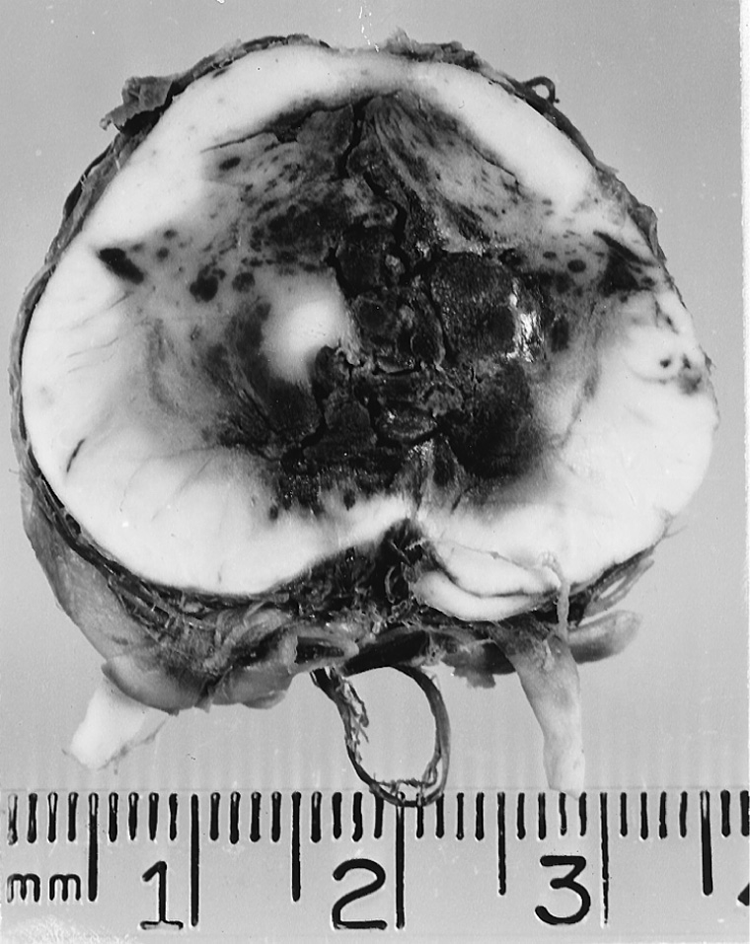
FIGURE 12-12. Transverse section of the mesencephalon showing hemorrhages secondary to transtentorial herniation. The IIIrd nerves are seen exiting ventrally. The caudal displacement of the brainstem by the herniating cerebrum stretches and compresses the brainstem vessels until they rupture, killing the patient.
J. Decerebrate rigidity, a postural syndrome of midbrain lesions
1. Description of the decerebrate posture: Extensive midbrain lesions, intrinsic or from unilateral or bilateral transtentorial herniation, disconnect the cerebrum and diencephalon from the rest of the brainstem, that is, decerebrate the individual. The “decerebration” can, in fact, be toxic–metabolic rather than anatomic. Along with the IIIrd nerve signs, double hemiplegia, and loss of consciousness, the Pt may show a diagnostic postural syndrome called decerebrate rigidity (gamma rigidity). Study Fig. 12-13 and assume the decerebrate posture yourself. Can you maintain it easily?
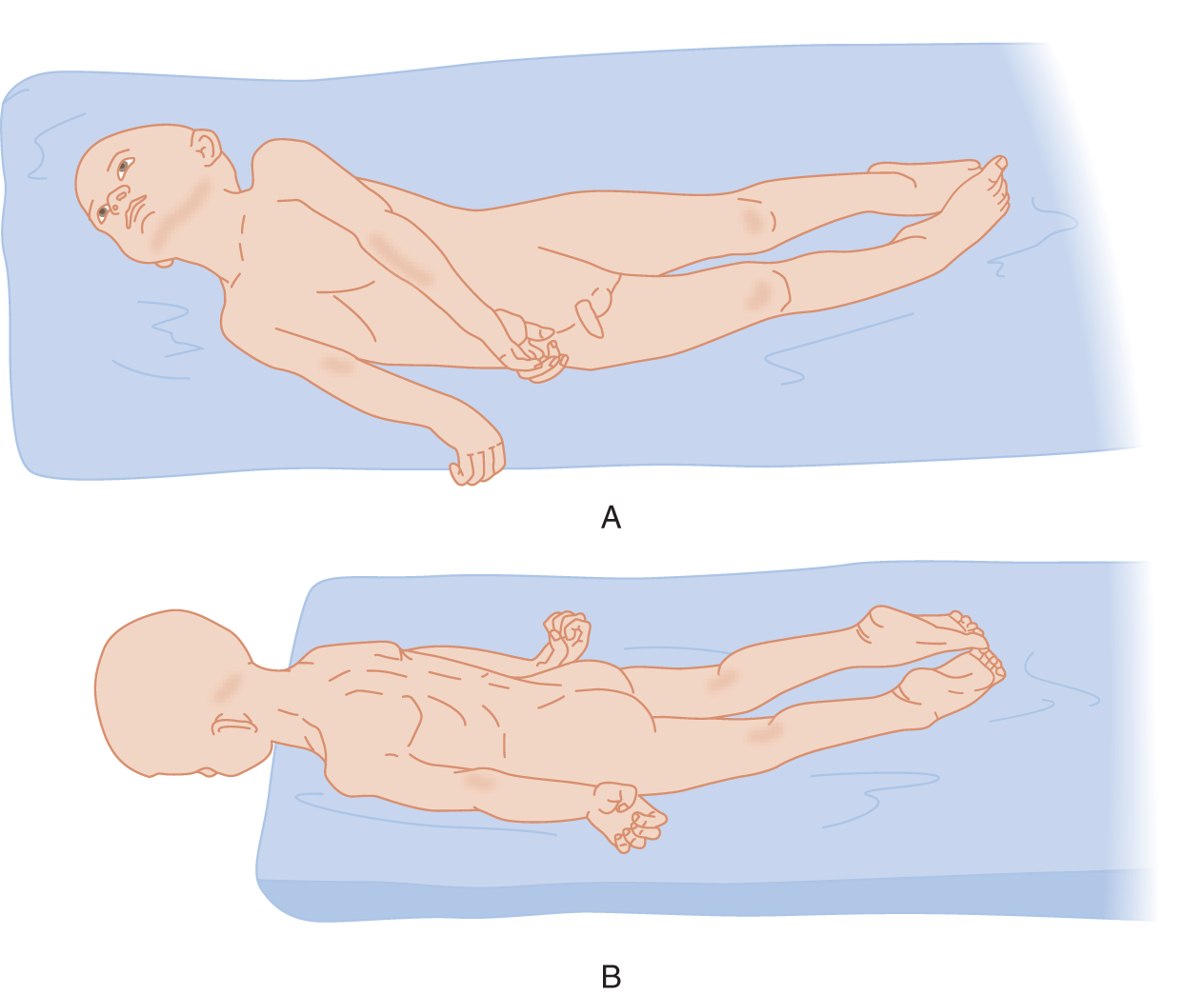
FIGURE 12-13. Decerebrate posture in an unconscious Pt. (A) Patient placed on his side. (B) Patient placed with his head over the edge of the table to show the rigid extension of his neck. (Adapted with permission from Penfield W, Jasper H. Epilepsy and the Functional Anatomy of the Human Brain. Boston, MA: Little, Brown & Co.; 1954).
2. After carefully inspecting the head, jaw, trunk, and limb postures of the Pt, cover Fig. 12-13 and test your powers of observation by completing Table 12-3.
TABLE 12-3 • Posture in Decerebrate Rigidity
The mouth is |
The head is |
The trunk is |
The arms are |
The wrists are |
The fingers are |
The legs are |
The feet are |
The toes are |
3. Muscle tone in decerebrate rigidity: Testing of the muscle tone of the Pt shown in Fig. 12-13 would disclose rigid extension of the extremities and spine, but if you once bend the part, it will yield like a clasp knife. Hence, the tonic disturbance seems to combine features of spasticity and rigidity.
4. Descriptive definition of decerebrate rigidity: The Pt displays an involuntary posture with the:
a. Proximal joints (spine, shoulders, hips, elbows, and knees) rigidly extended/ flexed. (extended)
b. Distal joints (wrists/ankles, fingers/toes) rigidly extended/ flexed. (flexed)
c. Forearms and legs internally/ externally rotated. (internally)
5. Pathophysiologic explanation of the decerebrate posture: As a prelude to interpret the decerebrate posture, review the strength of the muscle actions listed in Table 12-4. Actually test a normal subject to complete the table.
TABLE 12-4 • Comparative Strength of Postural Deviation in Decerebrate Rigidity
Action |
Stronger than |
Weaker than |
Opposing action |
Jaw closure is |
|
|
Jaw opening |
Head extension is |
|
|
Head flexion |
Trunk extension is |
|
|
Trunk flexion |
Arm extension is |
|
|
Arm flexion |
Forearm pronation, arm extended is |
|
|
Supination |
Wrist flexion is |
|
|
Wrist extension |
Finger flexion is |
|
|
Finger extension |
Leg extension is |
|
|
Leg flexion |
Inversion of foot is |
|
|
Eversion |
Ankle flexion is |
|
|
Ankle extension |
Toe flexion is |
|
|
Toe extension |
a. After completing Table 12-4, you can conclude that the direction of pull of the strongest muscles determines the posture of the spine and extremities assumed by the decerebrate Pt.
b. Figure 12-14 shows the posture of a quadruped with decerebrate rigidity. A quadruped with decerebrate rigidity will stand, if set on its feet. Review also Fig. 7-2.
c. The extended head, tail, and extremities and the closed jaw of the quadruped indicate that the muscles that acting to cause the decerebrate posture support a quadruped animal’s posture against collapse due to the pull of gravity. These same muscles cause it to leap and locomote in defiance of gravity. Hence, we interpret decerebrate rigidity as that posture maintained by excessive contraction of the antigravity muscles of the quadruped. In general, the strongest muscles of humans are the true antigravity muscles of a quadruped/biped. (quadruped (Review Table 12-3 and Fig. 7-2, if you missed.))
d. Describe how you could produce the decerebrate posture in an experimental animal._________

FIGURE 12-14. Decerebrate posture in the cat. (Reproduced with permission from Pollock L, Davis L. The reflex activities of a decerebrate animal. J Comp Neurol. 1930;50:377–411.)
6. Role of the vestibular system in decerebrate rigidity: After decerebration by midbrain transection interrupts the impulses descending from the cerebrum, the vestibular system becomes overactive, causing it to overstimulate or drive the powerful antigravity muscles. (If you tested carefully, you should have checked the stronger than column every time.)
a. Explain whether you should classify the decerebrate posture as a deficit or a release phenomenon._________
b. Describe a surgical experiment or series of experiments to prove that the drive for the decerebrate posture comes from the vestibular system. Think carefully.
_________
_________
c. Like any release phenomenon, decerebrate rigidity not only requires a lesion to release it but also an intact neural mechanism to produce, drive, and perpetuate it. The term “decerebrate rigidity” was first used by Sherrington (1898) after an intercollicular transection (between the red nuclei and the vestibular nuclei) in animal studies. Sherrington showed that destruction of the vestibular system abolishes the decerebrate posture. Apparently the activity that drives the decerebrate posture arises in the vestibular system and is conveyed to the spinal cord by the vestibulospinal tracts. Dorsal or ventral root transection also abolishes it.
d. For decerebrate rigidity to appear, the critical neural structure transected or compressed is the _________
7. Give a succinct clinical description of the decerebrate posture. Be sure to separate observation from interpretation._________
_________
8. Give the pathophysiologic interpretation of decerebrate rigidity.
_________
_________
9. Some unconscious Pts show decerebrate posturing only in response to pain. To elicit the posture, press the ball of your thumb or a knuckle hard for several seconds against the Pt’s sternum.
10. Despite the relatively poor prognosis, rarely Pts with decerebrate rigidity remain conscious (Halsey and Downie, 1966) and rarely recover (Conomy and Swash, 1968; Brendler and Selverstone, 1970; Damasceno, 1991; Kao et al, 2006).
K. Decorticate versus decerebrate posture
1. The decerebrate posture differs from chronic hemiplegia (after the state of cerebral shock or diaschisis in acute hemiplegia has passed). Compare Figs. 12-13 and 12-15.

FIGURE 12-15. Posture in an adult with chronic hemiplegia. Compare the arm position with that shown in Fig. 12-13.
2. Describe the major difference in the posture of the arm in decerebrate rigidity versus hemiplegia.
_________
_________
L. Respiratory and autonomic effects of brainstem lesions and transtentorial herniation
1. The unconscious Pt may display strong inspiratory stridor because of collapse of the oropharyngeal muscles. Simply place your fingers behind the ramus of the mandible and pull the mandible forward. The Pt will immediately breathe more freely. Positioning the head to the side or placing an oropharyngeal airway then solves the problem.
2. Lesions at various levels from the cerebrum to the upper cervical cord cause characteristic respiratory dysrhythmias. Figure 12-16 depicts some of the dysrhythmias, but it depicts their localizations more schematically than the data warrant.
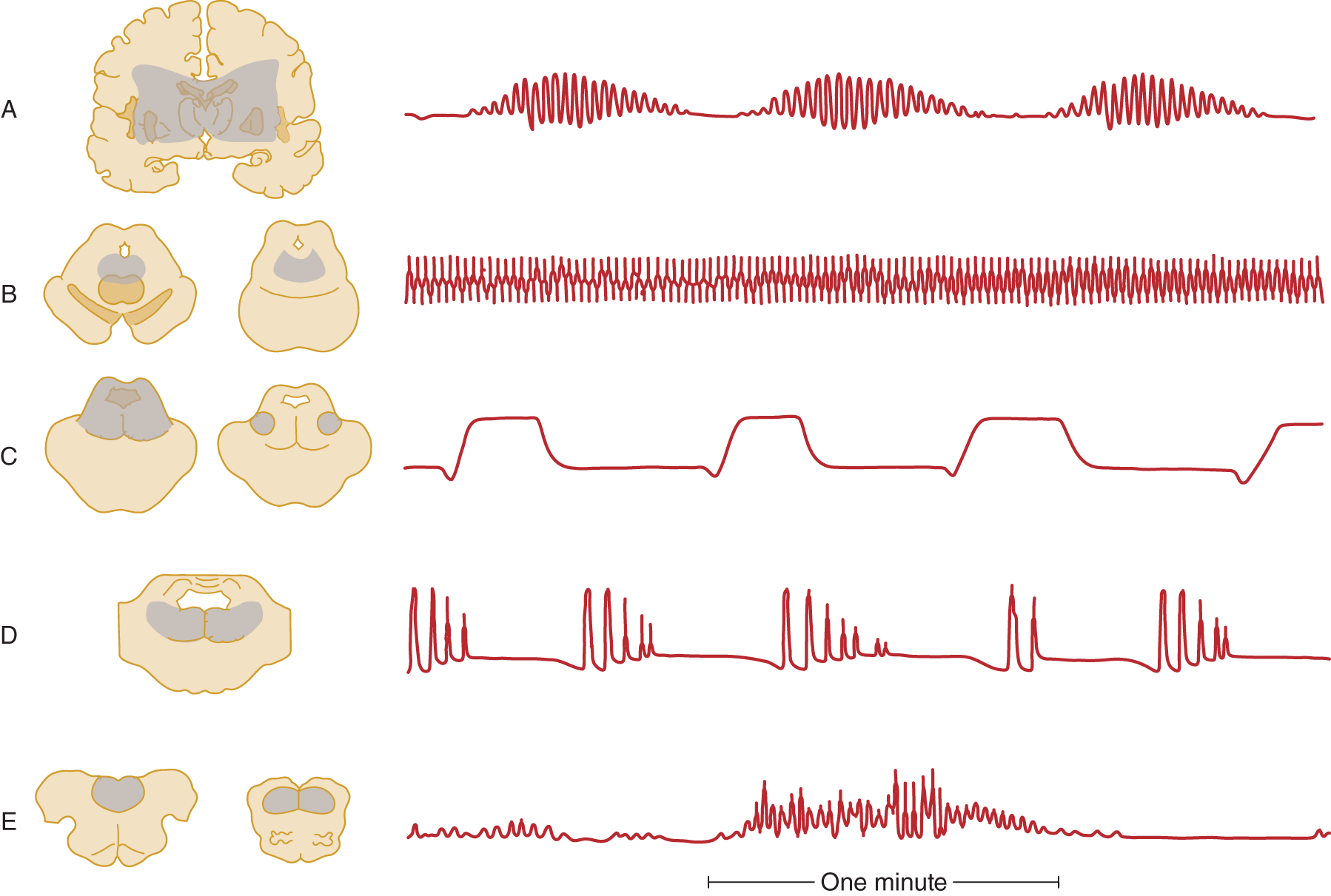
FIGURE 12-16. Correlation of intra-axial brainstem lesions at successive levels, with the type of respiratory dysrhythmia caused. (A) Cheyne–Stokes respiration. (B) Central neurogenic hyperventilation. (C) Apneustic breathing. (D) Cluster breathing. (E) Ataxic breathing. (Redrawn from Plum F, Posner J. The Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.)
3. Transtentorial herniation, by external compression, caudal displacement, and kinking of the brainstem, causes a “wave of failure” to pass caudally from diencephalon to medulla. The Pt passes in succession through the various respiratory dysrhythmias shown in Fig. 12-16 that reflect the effects of lesion at various rostrocaudal levels. One type of dysrhythmia seen with intrinsic brainstem lesions but not with transtentorial herniation is apneustic breathing (Fig. 12-16C; Plum and Posner, 1980).
4. Among many other dysrhythmias not specific as to lesion site or cause are Biot and Kussmaul.
a. Kussmaul breathing is characterized by “air hunger,” with deep regular sighing respiration (increase tidal volume), whether the rate is slow, normal, or fast. Kussmaul respiration suggests the presence of metabolic acidosis, and is the result of stimulation of the brainstem respiratory centers by the low blood pH. It occurs with metabolic disorders, such as diabetic ketoacidosis and uremia, exogenous acids (eg, intoxication with salicylate methanol, or ethylene glycol), and other severe systemic illnesses.
b. Biot breathing resembles Cheyne–Stokes breathing (Fig. 12-16A) by consisting of periods of apnea that alternate irregularly with series of breaths, but the four to five breaths have equal length and terminate abruptly rather than fading in and out.
5. The effect of transtentorial herniation or other brainstem lesions on blood pressure and pulse varies. Although hypertension or hypotension and tachycardia or bradycardia may occur, typically increased ICP causes a slowing of the pulse rate and an increasing blood pressure (Cushing phenomenon). Many factors, such as the ventilatory efficiency, may alter this formula. Thus, the Ex must expect changes in the vital signs and monitor them carefully.
M. Transforaminal herniation
1. Anatomy of transforaminal herniation: The diffusely swollen, herniating brain may impose the death penalty by a mechanism other than transtentorial herniation. As increased pressure pushes the intracranial contents caudally, the cerebellum and medulla herniate into the foramen magnum. This aperture is designed to accommodate only the cervicomedullary junction.
a. Because the caudalmost part of the cerebellum, the cerebellar tonsils, is the part of the cerebellum to herniate, this lesion is called tonsillar or transforaminal herniation. See the herniated right cerebellar tonsil shown in Fig. 12-8.
b. Transforaminal herniation may result from expanding supra tentorial lesions or from expanding infratentorial lesions, such as a cerebellar hemorrhage or neoplasm. Chapter 13 describes the danger of transforaminal herniation after lumbar puncture (LP).
c. Infratentorial masses also may cause upward transtentorial herniation, causing midbrain compression and midbrain signs (Cuneo et al, 1979).
2. The clinical syndrome of transforaminal herniation: The clinical syndrome mimics transection of the neuraxis at the medullocervical junction level. The Pt becomes quadriplegic and totally apneic (Table 12-5). The total apnea results from the quadriplegia that stops volitional breathing and compression of the reticulospinal tracts that stops automatic breathing. No respiratory drive reaches the lower motoneurons (LMNs). Review Fig. 6-17.
TABLE 12-5 • Summary of Vascular Complications and Clinical Signs of Internal Herniations of the Brain
Type of herniation |
Vascular complications |
Clinical signs |
Subfalcine herniation medially of the cingulate gyrus |
Compression of anterior cerebral artery under free edge of the falx, causing infarction of medial hemispheric wall and motor area for the leg (Figs. 2-2C and 12-9) |
Obtundation, disorientation, monoparesis of contralateral lower extremity |
Transtentorial herniation downward and medially of the uncus and parahippocampal gyrus |
Compression of posterior cerebral artery against free edge of tentorium (Figs. 12-6 and 12-7); brainstem hemorrhages (Fig. 12-12). The anterior choroidal and posterior communicating arteries may be displaced inferomedially in severe cases |
Increasing loss of consciousness, pupillodilation; and IIIrd nerve palsy; hemianopia |
Transtentorial herniation upward of the cerebellar vermis and midbrain |
Compression of superior cerebellar artery by free edge of tentorium and compression of midbrain and brainstem vessels |
Increasing obtundation, pretectal/rostral midbrain syndrome (Table 5-2), IIIrd nerve palsy |
Transforaminal herniation downward of the cerebellar tonsils and medulla oblongata |
Infarction of the cervicomedullary junction and rostral 1-2 segments of spinal cord from compression of ventral and posterior spinal arteries that arise inside the foramen magnum from the vertebral and posterior inferior cerebellar arteries; hemorrhagic necrosis of the cerebellar tonsils |
Neck stiffness or head tilt. Apnea and quadriparesis from compression of the medullocervical junction |
N. Summary of the internal herniations of the brain
1. Name the three common internal herniations of the brain.
_________
2. Name the parts of the brain that herniate in transtentorial herniation and in transforaminal herniation. _________
_________
3. State, in principle, how brain herniations kill the Pt.
_________
_________
_________
4. Could you test for cerebellar function in a comatose Pt? Yes/ No. Explain.
_________
_________
_________No) (Clinical tests for cerebellar dysfunction require conscious responses by the Pt.)
5. If a comatose Pt removed from a wrecked car shows distinct decerebrate rigidity in the upper extremities and the lower extremities are totally flaccid, what would be the best clinical conclusion? Think circuitry to figure out and explain the answer.
_________
_________
6. The Pt may have suffered spinal cord transection and a brain injury, a not uncommon association. In this case, a single lesion does not explain the clinical picture, but a single cause, trauma, does. Sometimes however, even with an intact spinal cord, only the arms will show decerebrate posturing. Hence, the action of the legs proves that the cord is intact, but the absence does not. Extremely rarely, posturing of the arms resembling decerebrate rigidity occurs as a spinal reflex (Saposnik et al, 2000; Brain Death bibliography).
O. Brain herniations: a clinical summation and critique
1. Table 12-5 summarizes the internal herniations. Cover the text in columns 2 and 3 and recite the text by using the headings in column 1 as a prompt.
2. When any modern gladiator, a boxer, a football player, or a race driver, dies after a head injury, in sacrifice to our appetite for brutality, you can surmise that the mechanism of death was brain edema and herniation (Jordan, 1992; Unterharnscheidt, 1970, 2003). If we, as a people, do not value above all else the preciousness and fragility of life, one might suppose that we would, at least, value and protect the brain, which gives us the means to seek advantages. For the brain functions not, as you might have supposed, as an organ of intelligence, but, as Szent–Györgyi said, as an advantage-seeking organ. The television hero stunned by a head injury who, without lingering effects, immediately returns to chasing the bad guys represents an egregious error in medical management because it trivializes head injury. Getting “dinged” is sports slang to minimize a possible life-taking event. Any person stunned or unconscious from a blow to the head requires rest and careful medical observation. You must presume that any Pt with any head injury may have a life-threatening epidural or subdural hematoma—albeit initially inapparent. Whether to send home the Pt who has had a seemingly trivial blow to the head or to hospitalize for observation is one of the most difficult of all medical decisions. You must deal with prediction, the most difficult of all the arts. Remember that the specter of death is looking over your shoulder.
Life is short, the art long, opportunity fleeing, experience treacherous, judgment difficult.
3. To carefully monitor every Pt who has a potential brain herniation, record the mental status and all signs: vital signs, such as blood pressure and pulse, and physical signs, such as the pupillary diameters in millimeters. Record the presence or absence of venous pulsations (Jacks and Miller, 2003). The goal always is to intervene early to prevent herniations. Given a Pt with a head injury and left hemiparesis who is initially conscious, outline the sequence of events that will predict transtentorial herniation and list the sequence of events to death:
a. Consciousness: _________
b. Extremity movement and posture: _________
c. Pupillary changes: _________
d. Respiration changes: _________
e. Pulse and blood pressure: _________
4. Answers to frame 3
a. May have some excitement or delirium and then pathologic sleep, stupor, and coma.
b. Left hemiparesis may worsen to hemiplegia, and right hemiparesis appears and leads to double hemiplegia, often followed by decerebrate rigidity and then complete flaccidity just preceding death as the brainstem becomes more and more damaged.
c. The right and then the left pupil become dilated and fixed. Both eyes become immobile.
d. Cheyne–Stokes, central neurogenic hyperventilation, cluster breathing, ataxic or Biot respiration, and total apnea.
e. Pulse and blood pressure may fluctuate, but there is a tendency for blood pressure to increase with decreasing pulse rate (Cushing phenomenon).
f. Hemorrhage into the brainstem, secondary to stretching of the brainstem vessels.
5. The time to help has essentially passed when the Pt shows the full signs of brain herniation, with deep coma, bilateral pupillodilation, and decerebrate rigidity. At that point, many Pts are irretrievable. The Ex must have recognized the Pt’s increased pressure and potential for herniation and called for neurologic help (Clusmann et al, 2001). If the physicians can relieve the pressure early, the Pt recovers; if not, the Pt dies. That is the importance of understanding the mechanisms and clinical signs described here.
BIBLIOGRAPHY · Internal Herniations of the Brain: Effect on Consciousness, Neurologic Function, and Vascular System
Brendler SJ, Selverstone B. Recovery from decerebration. Brain. 1970;93:381–392.
Clusmann H, Schaller C, Schramm J. Fixed and dilated pupils after trauma, stroke, and previous intracranial surgery: management and outcome. J Neurol Neurosurg Psychiatry. 2001;71:175–181.
Conomy JP, Swash M. Reversible decerebrate and decorticate postures in hepatic coma. N Engl J Med. 1968;278:876–879.
Cuneo RA, Caronna JJ, Pitts L, et al. Upward transtentorial herniation: seven cases and literature review. Arch Neurol. 1979;36(10):618–623.
Damasceno BP. Decerebrate rigidity with preserved cognition and gait: a possible role of anoxic-ischemic brain damage. Int J Neurosci. 1991;58:283–287 (Medline).
Davis RL, Robertson DM, eds, Textbook of Neuropathology. 2nd ed. Baltimore, MD: Williams and Wilkins; 1991.
Fisher CM. Brain herniation: a revision of classical concepts. Can J Neurol Sci. 1995;22:83–91.
Forbes H, Norris JR, Fawcett J. A sign of intracranial mass with impending uncal herniation. Arch Neurol. 1965;12(4):381–386.
Friede R, Roessmann U. The pathogenesis of secondary midbrain hemorrhages. Neurology. 1966;16:1210–1216.
Halsey J, Downie A. Decerebrate rigidity with preservation of consciousness. J Neurol Neurosurg Psychiatry. 1966;29:350–355.
Hassler O. Arterial pattern of human brainstem: normal appearance and deformation in expanding supratentorial conditions. Neurology. 1967;17:368–375.
Inao S, Kawai T, Kabeya R, et al. Relation between brain displacement and local cerebral blood flow in patients with chronic subdural haematoma. J Neurol Neurosurg Psychiatry. 2001;71:741–746.
Jordan B. Medical Aspects of Boxing. Boca Raton, FL: CRC Press; 1992.
Kao CD, Guo WY, Chen JT, et al. MR findings of decerebrate rigidity with preservation of consciousness. AJNR. 2006;27:1074–1075.
Keane JR. Blindness following tentorial herniation. Ann Neurol. 1980;8:186–190.
Kirschner HS, Staller J, Webb W, et al. Transtentorial herniation with posterior cerebral artery territory infarction: a new mechanism of the syndrome of alexia without agraphia. Stroke. 1982;13:243–246.
Plum F, Posner J. The Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.
Pollock L, Davis L. The reflex activities of a decerebrate animal. J Comp Neurol. 1930;50:377–411.
Pullicino PM, Alexandrov AV, Shelton JA, et al. Mass effect and death from severe acute stroke. Neurology. 1997;49:1090–1095.
Ropper AH, ed. Neurological and Neurosurgical Intensive Care. 3rd ed. New York, NY: Raven Press; 1993.
Ropper AH. The opposite pupil in herniation. Neurology. 1990;40:1707–1709.
Saeki N, Higuchi Y, Sunami K, et al. Selective hemihypaesthesia due to tentorial coup injury against the dorsolateral midbrain: potential cause of sensory impairment after closed head injury. J Neurol Neurosurg Psychiatry. 2000;69:117–118.
Sherrington CS. Decerebrate rigidity and reflex coordination of movements. J Physiol. 1898;22:319–322.
Sunderland S. The tentorial notch and complications produced by herniations of the brain through that aperture. Br J Surg. 1958;45:422–438.
Unterharnscheidt F. About boxing: review of historical and medical aspects. Texas Rep Biol Med. 1970;28:421–425.
Unterharnscheidt F, Unterharnscheidt JT, eds. Boxing Med Aspects. San Diego, CA: Academic Press; 2003.
Walker A. The syndromes of the tentorial notch. J Nerv Ment Dis. 1963;136:118–129.
IV. INITIAL NEUROLOGIC EXAMINATION OF THE UNCONSCIOUS PATIENT
A. Introduction
1. For the most difficult diagnostic challenge of all, we would ask you to evaluate a Pt brought in comatose off the street, with no history available. Review the neurologic examination of the unconscious patient on page XX, at the front of the text. Recite the ABCDEE mnemonic, the 5H mnemonic for enemies of the brain (hint: start with Hypoxia), and review the differential diagnostic dendrogram. At your leisure, read Fisher (1995) and Posner et al (2007) for masterful descriptions of the neurologic examination (NE) of the unconscious Pt.
2. In trying to determine whether the putatively unconscious Pt is arousable, the inexperienced Ex seems instinctively to want to move the Pt’s head from side to side, shake a limb, or jar the gurney. Avoid these maneuvers until you can be sure that the Pt does not have a broken neck or a broken arm.
3. The seemingly unresponsive Pt may have a toxic or metabolic state, an anatomic lesion, trauma, or a mental illness. The Ex has to determine immediately whether the Pt has an anatomic lesion or a metabolic or toxic disorder that threaten life (Fig. NE-2). Asymmetric neurologic signs, such as a hemiparesis or a cranial nerve palsy, provide the best evidence of an anatomic lesion as the cause for the coma. The mental disorders get sorted out in the course of the evaluation for life-threatening disorders. Mental illnesses include somatization disorder, malingering, and psychosis with catatonia (catatonia consists of catalepsy or waxy flexibility, negativism, mutism, and bizarre posturing).
B. Inspection of the comatose patient
1. If the Pt can breathe adequately, so can the Ex (Video 12-2). The Ex has at least a little time for contemplation. Proceed to look for what is “good” and favorable and what is “bad” and evil. Anything that works normally, that is, breathing and pupillary light reflexes, is good. What does not work at all is evil. Therefore, we extract a “cosmic law” about the NE of the unconscious Pt. Whatever behavior the Pt shows, a pupil constricting or a limb moving, establishes the integrity of some neuroanatomic circuit and the function of some intact neurophysiologic mechanism (Videos 12-3 and 12-4).

Video 12-2. Impaired breathing associated with pseudocholinesterase deficiency in a patient who had inguinal hernia surgery.

Video 12-3. Base of the skull fracture resulting in raccoon’s eyes and a right Battle sign.

Video 12-4. Posttraumatic fourth nerve (CN IV) palsy.
2. If the Pt fails to make any responses, the Ex has to decide whether this results from anatomic interruption of central neuroanatomic circuits, the depth of the coma, or total effector paralysis, as in severe GBS or the Pt curarized for management on a respirator.
C. “Good” behaviors disclosed by the four-glance, instant screening neurologic examination of the acutely unconscious patient
Given a Pt who is unconscious (but not comatose, with no reflexes or behavior at all), the Ex can complete a preliminary but surprisingly adequate four-glance survey of the Pt’s neuroanatomic circuits. The value of the observations depends on your ability to “think circuitry” and the implications of the behavior observed.
1. Normal breathing and oropharyngeal reflexes: Breathing and related reflexes, such as coughing, swallowing, hiccuping, or yawning, establish that CrNs IX, X, and XII, the pontomedullary reticular formation, and cervical and thoracic levels of the spinal cord are intact and cannot be the site of a lesion (Fig. 6-17).
2. Blinking or tonic closure of the eyelids: CrNs V and VII work.
3. Random slow conjugate drifts of the eyes to the sides: CrNs III, IV, and VI and the frontopontine pathway that drives eye movements are intact (Fig. 2-30). Together with the integrity of CrNs V and VII, the entire pontine tegmentum is intact, because the nucleus of CrN V is in the rostral part of the pontine tegmentum, the nuclei of CrNs VI and VII are in the caudal part, and the medial longitudinal fasciculus extends the entire length. Given the drifts of the eyes, the same considerations prove that the midbrain is intact: the nucleus of CrN III is in the rostral midbrain tegmentum, that of CrN IV is in the caudal, and the medial longitudinal fasciculus (MLF) extends the entire length.
4. Random spontaneous, particularly semi-purposive symmetrical movements of all four extremities: Both pyramidal tracts, from the motor cortex to the sacral level of the spinal cord, work (Fig. 2-27). At this point, the Ex knows that the entire length of the neuraxis, from the cerebral cortex to the tip of the spinal cord, is more or less intact. If after the four glances, you then find intact pupillary, corneal, vestibular, and auditopalpebral reflexes, further demonstrating the functional integrity of the midbrain, pons, medulla, and their cranial nerves, you have completed a cerebral cortex to sacral cord screening NE that virtually excludes a large anatomic lesion as the cause for the unconscious episode.
5. In addition to random or semi-purposive movements, the unconscious Pt may show myokymia and a variety of mutterings, shivers, shudders, twitches, and myoclonias difficult to classify, in addition to more patterned movements of choreiform, athetoid, or dystonic type; or aimless picking at the bedclothes, a sign called carphologia or floccillation. These actions are of uncertain localizing or prognostic import but generally require intact pyramidal tracts.
6. In the differential diagnosis of coma, recall that active pulsations of the retinal veins almost exclude the presence of increased ICP. In fact, spontaneous venous pulsations disappear when the ICP as measured by lumbar puncture exceeds 190 mm of water (Levin, 1978). The presence of pulsations means more than their absence, because some normal persons do not show pulsations (Jacks and Miller, 2003). To best visualize venous pulsations requires direct ophthalmoscopy.
7. During this quick inspection, you may encounter more or less diagnostic odors of alcohol, fetor hepaticus, diabetic ketosis, or lung gangrene. The odor of alcohol does not establish it as the cause for the coma; perhaps the Pt has a subdural or epidural hematoma.
8. Also during the preliminary stage, note the Pt’s body temperature; hyperthermia and hypothermia suggest different categories of diagnosis.
D. “Bad” behaviors disclosed by inspection of the unconscious patient
1. Worst of all is no behavior whatsoever: flaccid eyelids and no pupillary responses; no eye drifts (in deep coma and brain death, the eyes return to and remain fixed in the neutral position); no breathing; no spontaneous movements; a totally flaccid, dumped-in-a-heap posture; and no response to any stimulus. At this point, the Ex does not know whether the absence of behavior results from destructive lesions, with neural shock or brain death, widespread LMN paralysis, or merely the depth of the coma, for example, as in a massive overdose of barbiturates.
2. Next worst is a sustained or driven posture: sustained deviation of the head and eyes or decerebrate rigidity.
a. Focal seizures may cause deviation of the head and eyes. In the absence of convulsions, you would expect a destructive lesion in the conjugate gaze center in the posterior frontal region of the ipsilateral/ contralateral hemisphere or in the ipsilateral/ contralateral half of the pons. (ipsilateral; contralateral (Review Figs. 5-1 and 5-3 if you missed.))
b. List which CrN(s) would most likely be paralyzed by a large lesion affecting the region of the pontine conjugate lateral gaze center._________
3. Prognostically bad findings: No prognostic formula unerringly predicts death, but Pts who fail to recover corneal and pupillary light reflexes within 24 hours of the onset of coma generally die. Similarly, Pts who have an acute head injury and who lack pupillary and vestibulo-ocular reflexes generally die. Prognostic scales have an error rate of 5% to 20% when applied to nontraumatic coma and require judicious interpretation to avoid premature termination of life support (Bates, 1991). The Ex should remain vigilant to the possibility of treatable metabolic derangements (eg, hypermagnesemia) mimicking herniation syndromes with fixed and dilated pupils (Rizzo et al, 1993)
E. Detecting hemiplegia in the unconscious patient
1. Inspection: The acute, severe anatomic lesions that cause unconsciousness or the hemiplegia that follows focal seizures (Todd paralysis) usually cause flaccid hemiplegia (cerebral shock). Look for unilateral asymmetry of movement, posture, muscle tone, and most importantly, unilateral flaccidity of the extremities. Unless deeply comatose, the unconscious Pt with intact pyramidal tracts moves all four extremities spontaneously or more or less purposively in response to pain. A tip-off posture of flaccid hemiplegia is that the leg rests in external rotation, with the foot turned out. Hemiplegia and a broken hip are the two most common reasons for this posture. Flaccidity on one side and absence of spontaneous or pain-induced movements identify acute hemiplegia. The face and extremities on the intact side continue to show some muscle tone.
2. Flaccidity of the cheek: When the unconscious hemiplegic Pt inhales, the cheek on one side sucks in; when the Pt exhales, that cheek puffs out. It will be on the hemiplegic/ nonhemiplegic side. Explain.
_________
_________hemiplegic) (Flaccid paralysis involves the buccinator and other facial muscles on the hemiplegic side. Therefore, the flaccid cheek on the hemiplegic side sucks in with inspiration and puffs out with expiration. The tone in the facial muscles on the other side holds the cheek in place.)
3. The eyelid-release test
a. Review of facial motor innervation
i. Diagram the UMN and LMN innervations of the right side of the face in Fig. 12-17. Check it against Fig. 6-3.

FIGURE 12-17. Blank to draw in the upper motoneuron innervation of the VIIth nerve nucleus and the lower motoneuron innervations of the face.
ii. Recall that the LMNs for the orbicularis oculi muscle may receive innervation by many crossed and uncrossed UMNs. Hence, in acute hemiplegia, after sudden interruption of UMNs, the eyelid may also show flaccidity and weakness. The eyelid-release test demonstrates this, because even in the unconscious (but not completely comatose) Pt, the eyelids remain closed because of tonic innervation, that is, by a positive drive.
b. Procedure for eyelid-release test: Gently pull both eyelids up with your two thumbs and then release them simultaneously (Fig. 12-18).
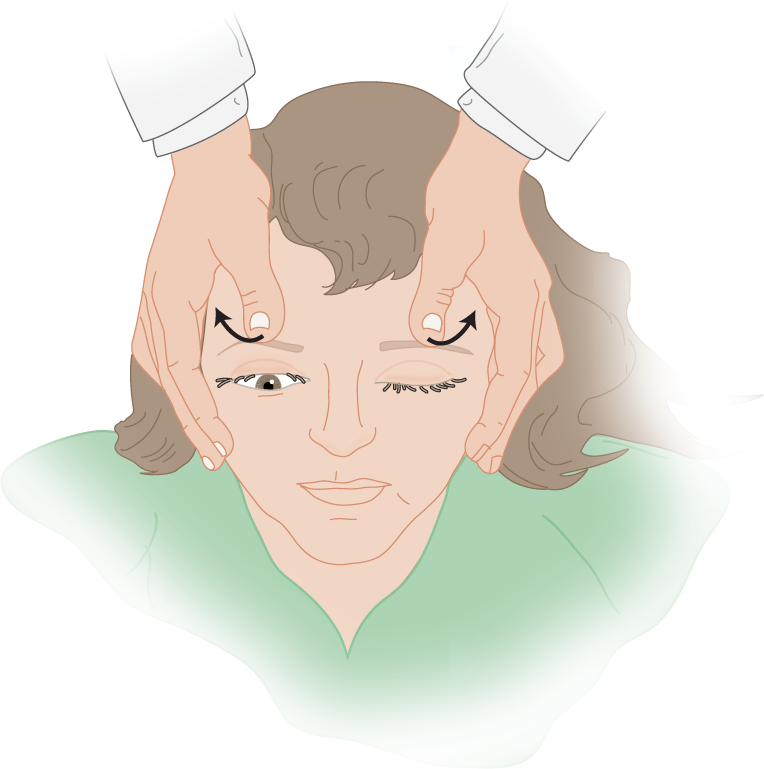
FIGURE 12-18. Eyelid-release test in a comatose patient with right hemiplegia. While standing at the head of the patient’s bed, the examiner elevates both lids and releases them simultaneously. The lid of the hemiplegic side closes slowly because of flaccidity of its orbicularis oculi muscle, whereas the lid of the normal side closes briskly because of tonus in its orbicularis oculi muscle.
c. Results: The eyelid of the hemiplegic side glides down slowly, whereas the opposite lid closes rapidly. In fact, it almost snaps shut, unless the Pt is deeply comatose. Why does the eyelid on the nonhemiplegic side close faster? _________
d. Rarely, the eyelids of the unconscious Pt will remain open and unblinking (Keane, 1975).
4. The limb-dropping tests: The limb-dropping tests demonstrate flaccid paralysis of the extremities.
a. The wrist-dropping test: Grasp both of the Pt’s forearms just proximal to the wrist. Hold the forearms vertical, as shown in Fig. 12-19. The flaccid hemiplegic wrist drops at right angles, whereas the nonhemiplegic wrist, having some tone, remains to some degree vertical.
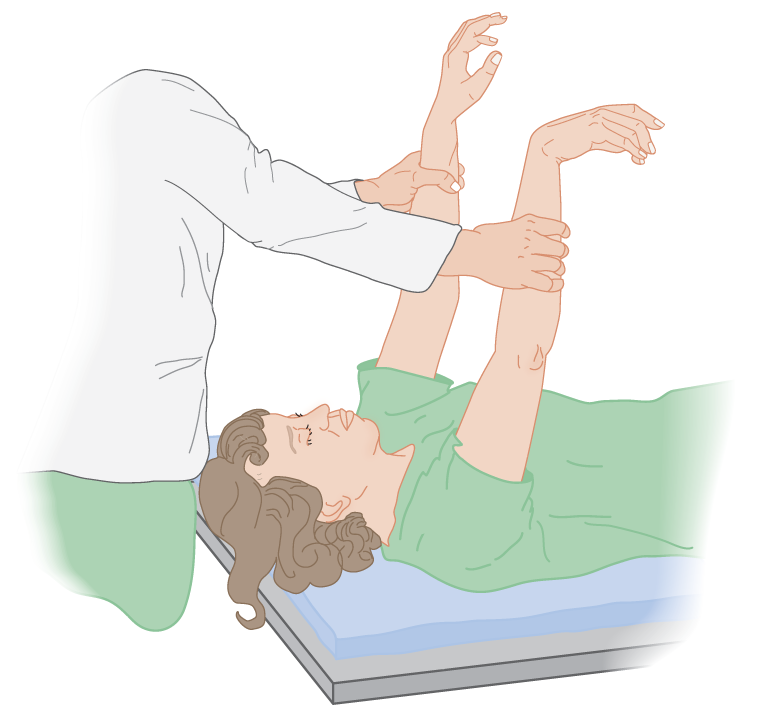
FIGURE 12-19. The wrist-dropping test for flaccid hemiplegia in a comatose patient with right hemiplegia.
b. The arm-dropping test: Grasp both forearms, as in the wrist-dropping test, and release them simultaneously. The hemiplegic arm drops limply, whereas the normal arm glides or floats down (Fig. 12-20). Lift the arm only a few inches and cushion its drop. Beware of ulnar nerve injury.
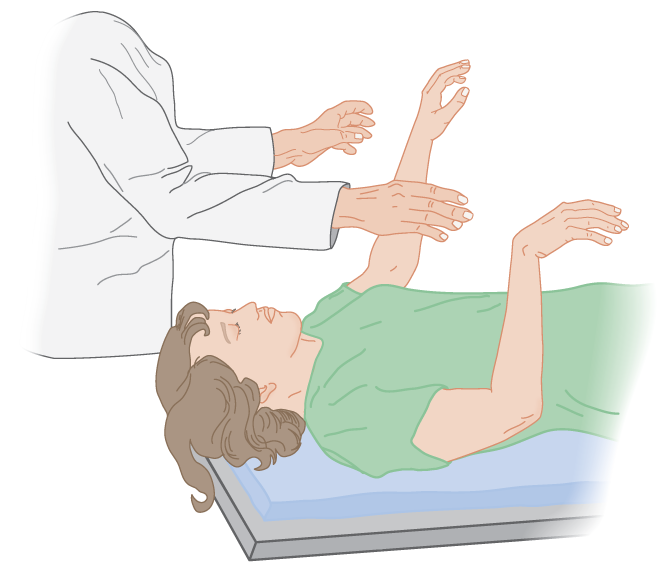
FIGURE 12-20. The arm-dropping test for flaccid hemiplegia in a comatose patient with right hemiplegia.
c. The leg-dropping test: Crook the Pt’s knees on your arm. Extend one leg first and drop it, and do the same with the other (Fig. 12-21). The Ex can both see and hear the difference as the flaccid hemiplegic leg drops more rapidly to strike the bed.
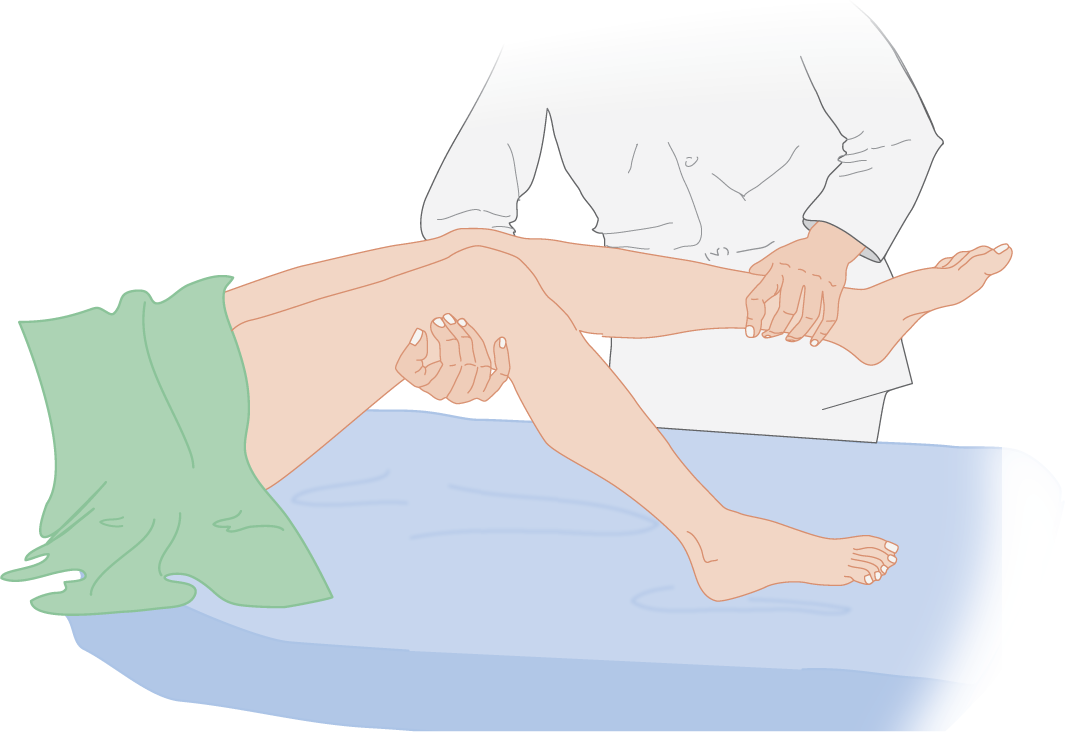
FIGURE 12-21. The leg-dropping test for flaccid hemiplegia in a comatose patient with right hemiplegia.
d. The dropping tests depend on the principle of asymmetry of muscle tone. For correct interpretation, the nonhemiplegic side must have some muscle tone. Hence, deep coma or LMN paralysis invalidates the tests.
5. Summarize the methods of detecting acute flaccid hemiplegia in an unconscious Pt._________
_________
F. Resistance to movement: paratonia (Gegenhalten)
1. The paratonic Pt may resist movement of a part of the body in any direction. It occurs in conscious and unconscious Pts. It is as if the Pt divines every movement you impose and automatically counteracts it.
2. Paratonia, like any resistance dependent on muscular contraction, does not occur on the side of acute flaccid hemiplegia or in the deeply comatose Pt when all tone is lost. The novice often misidentifies the nonhemiplegic side as hemiplegic because of mistaking paratonia for the increased tone of a UMN lesion.
G. Resistance to movement: nuchal rigidity and meningeal irritation signs
1. Definition of nuchal rigidity: The term nucha refers to the back of the neck. Nuchal rigidity means that neither the Pt nor the Ex can flex the Pt’s head because of reflex spasm of the nuchal (extensor) muscles. Irritation of the subarachnoid space, most commonly by inflammation (encephalitis or meningitis) or by subarachnoid blood, causes nuchal rigidity.
2. Suspension of the spinal cord: The spinal cord is buoyed by the surrounding CSF and suspended in position by its attachment to the medulla, by its nerve roots, and by special suspensory ligaments, the denticulate ligaments (Emery, 1967; Fig. 12-22).
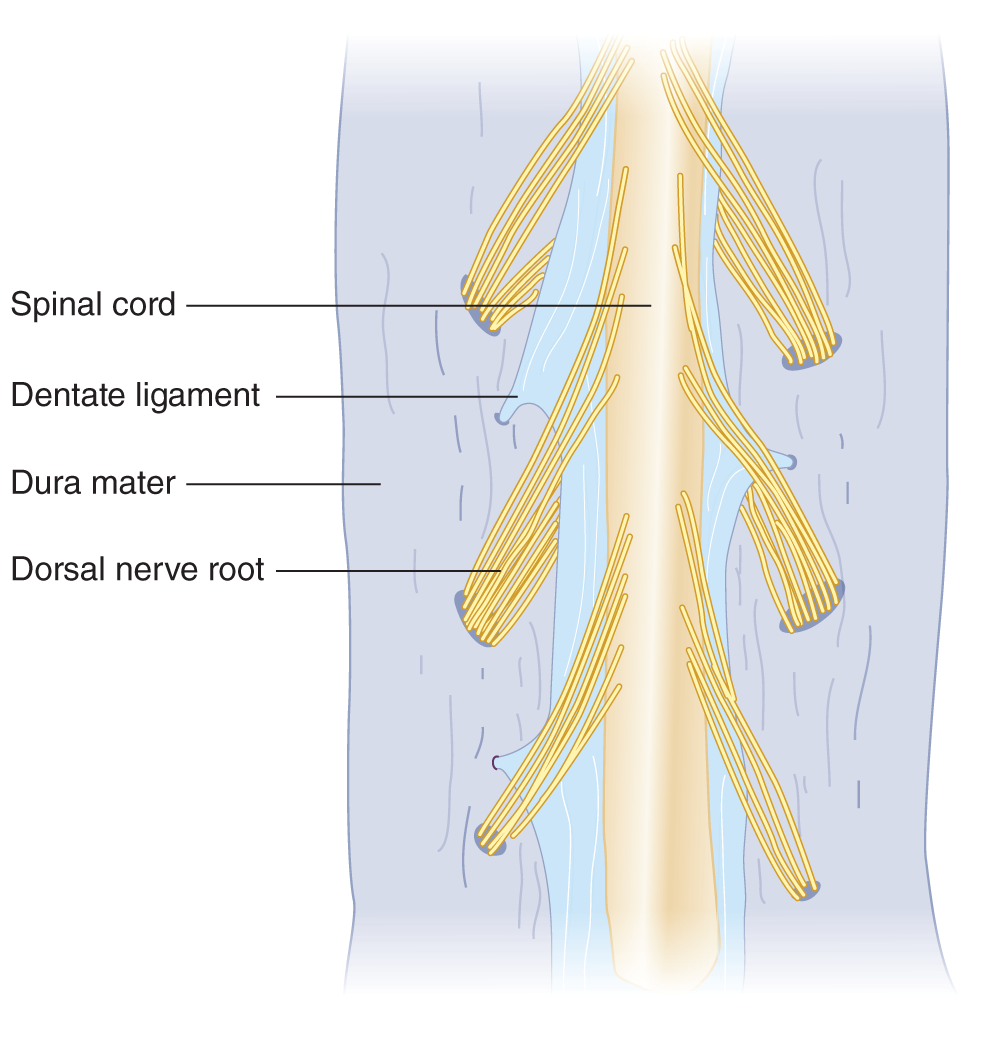
FIGURE 12-22. Dorsal view of the spinal cord, with the dura split open. Notice the dentate (denticulate) ligaments that suspend the spinal cord.
3. Biomechanics of neck flexion and extension: By understanding the mechanics involved in eliciting nuchal muscle spasm, you get a bonus: you learn how to position a Pt who has suffered a vertebral fracture and how to interpret Lhermitte sign. Study Figs. 12-23 and 12-24.
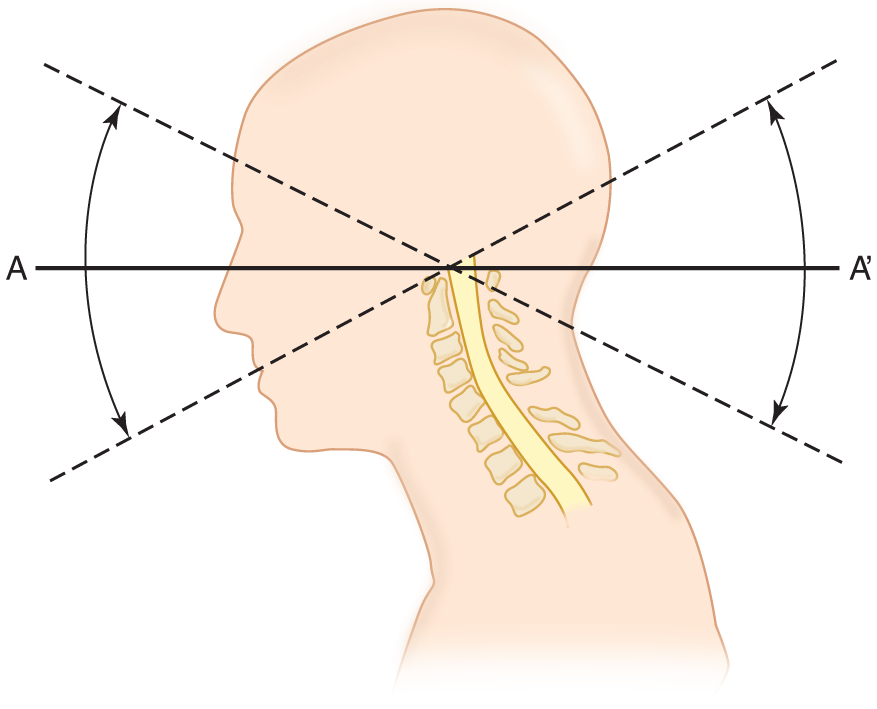
FIGURE 12-23. Sagittal section of the head to show the effect of head extension and flexion in stretching and relaxing the spinal cord. Imagine a steel rod driven through the head (A- A′). It acts as a lever with the fulcrum over the body of the first cervical vertebra.
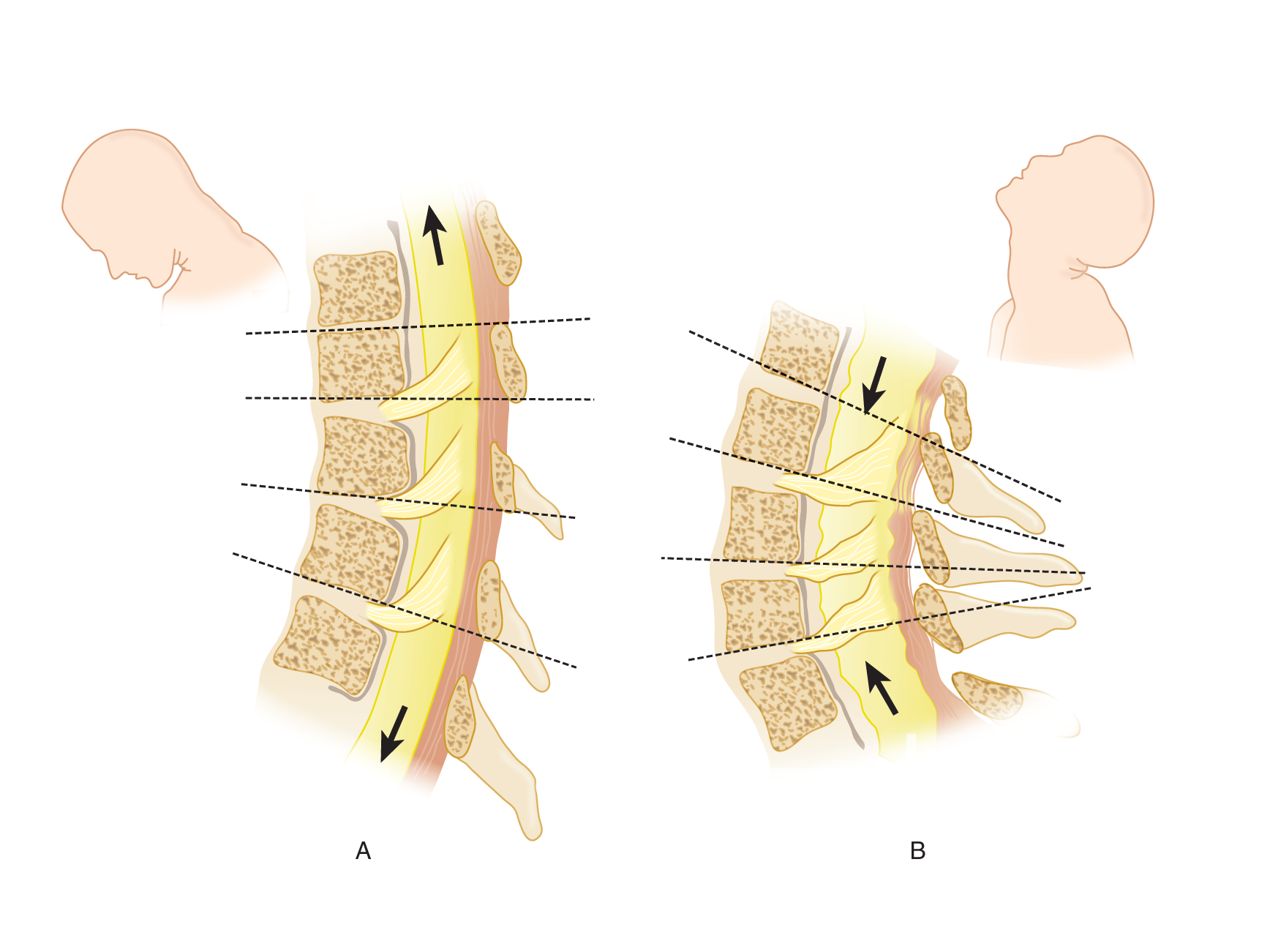
FIGURE 12-24. Sagittal sections through the cervical spinal cord and vertebrae. (A) Head flexion. (B) Head extension. Notice that the spinal cord and nerve roots are stretched and lengthened in A and relaxed and pleated in B.
a. Line A–A′ in Fig. 12-23 impales the neuraxis. It simulates a lever with the fulcrum at the top of the vertebral column. Thus, flexion of the head causes relaxation/ stretching of the spinal cord. (stretching)
b. During neck flexion and extension, movement occurs not only between the skull and first cervical vertebra but also between all other cervical vertebrae. The neck does not bend as though hinged at one point: It bends in a curve, like a sapling. The interrupted lines in Figs. 12-24A and 12-24B show the actual changes in vertebral angulation with flexion and extension and the accompanying changes in tension on the nerve roots and spinal cord (Breig, 1978; Goel and Weinstein, 1989).
4. The mechanism of spasm of the nuchal (extensor) muscles: The pain–spasm–pain cycle
a. During the range of movement from neck extension to flexion, the cord and roots change from a relaxed accordion-like wrinkling to a stretched condition (Fig. 12-24; Breig, 1978). When, however, roots, meninges, and cord are inflamed and swollen, the ordinarily innocuous act of flexing the head puts tension on the inflamed structures. It is like pressing on a boil. As long as the part with the boil remains still, it does not hurt. However, movement of the part that places tension on the swollen tissues causes intense pain. Any pain of this type elicits immediate muscle spasm, which prohibits the painful movement (O’Connell, 1946; Wartenberg, 1950). If you have ever tried to take a deep breath when you had a “stitch” in your side from a pleuritic rub, you will know how effectively pain inhibits the muscular contraction that causes it. Other examples of pain-avoiding muscle spasm are the abdominal wall rigidity of peritonitis and back rigidity from a lumbar sprain, that is, a “crick in the back.”
b. With inflamed meninges, the position that causes pain from tension on the cord and nerve roots is neck extension/ flexion. (flexion)
H. Technique to test for nuchal rigidity
1. With the Pt supine and relaxed, place your hand under the Pt’s occiput and gently attempt to flex the neck. Normally, it bends freely. If the Pt has nuchal rigidity, the neck resists flexion and the Pt winces with pain. If nuchal rigidity is severe, you can lift up the Pt’s head and trunk as if the spine were a rigid rod or as if the Pt were a statue (Fig. 12-25A).
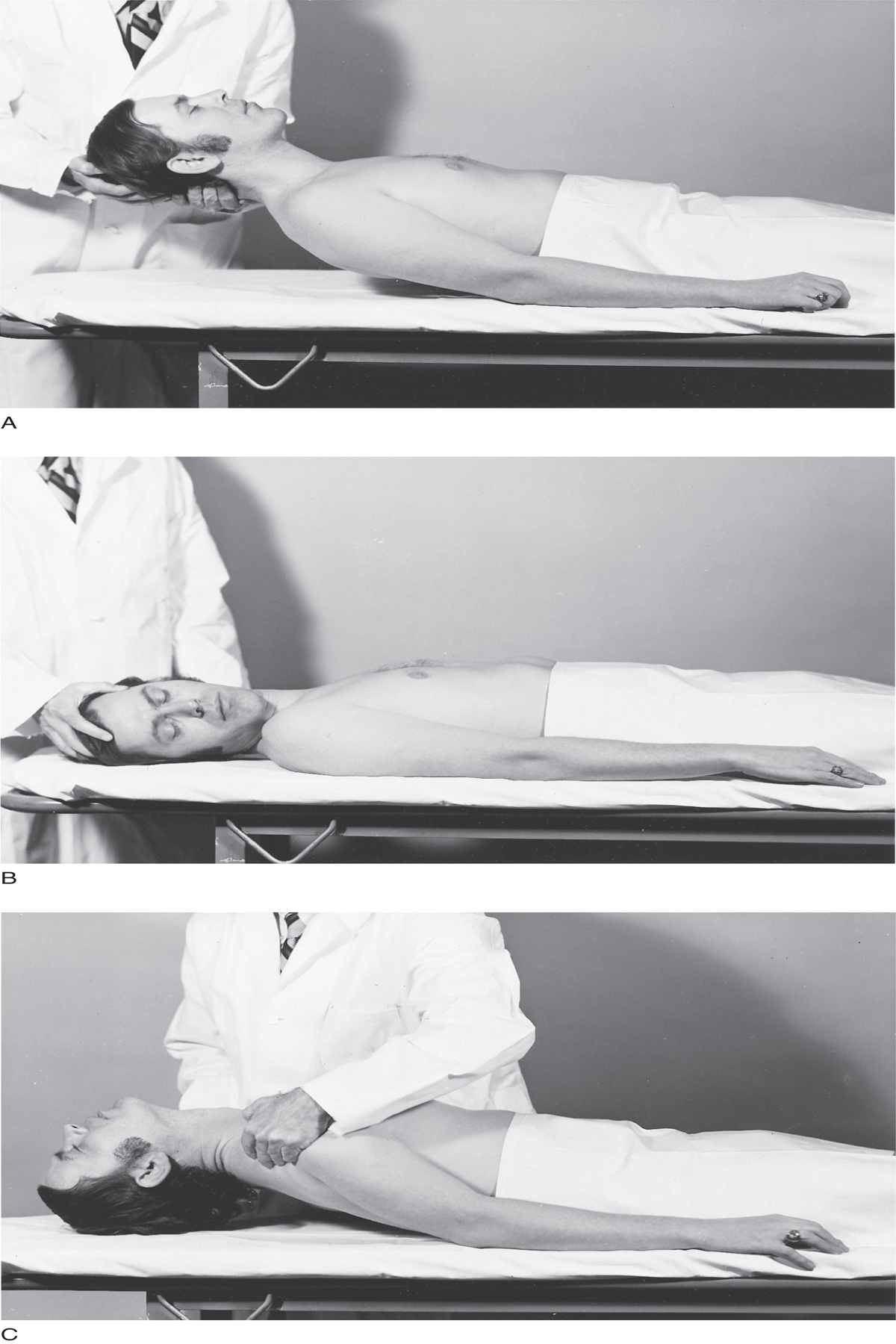
FIGURE 12-25. Diagnosis of pure nuchal rigidity, indicative of meningeal irritation. (A) The neck strongly resists flexion, often to the degree that the examiner can actually pick up the patient’s head and spine like a statue. (B) The neck rotates freely despite extreme nuchal rigidity that resists flexion. (C) The head falls back freely when the examiner lifts the patient’s shoulders.
2. Because true nuchal rigidity indicates meningeal irritation, the Ex must distinguish it from other forms of cervical rigidity. With true nuchal rigidity, the neck resists only flexion. The neck moves freely through rotation and extension, because these movements do not stretch meninges, spinal cord, and nerve roots. To demonstrate that rigidity affects only the nuchal muscles, do the following two things:
a. Place your hand on the Pt’s forehead. Passively roll the Pt’s head from side to side to demonstrate free head rotation despite resistance to flexion (Fig. 12-25B).
b. Then lift the Pt’s shoulders to let the head fall backward, testing for freedom of extension (Fig. 12-25C).
c. Cervical rigidity means any resistance to neck movement in any direction. In contrast, nuchal rigidity specifically means resistance to neck flexion, that is, rigidity of the back/ front of the neck. Figure 12-26 lists some of the numerous causes of cervical rigidity (Hunderfund et al, 2008; O’Connell J, 1946). (back (nape))
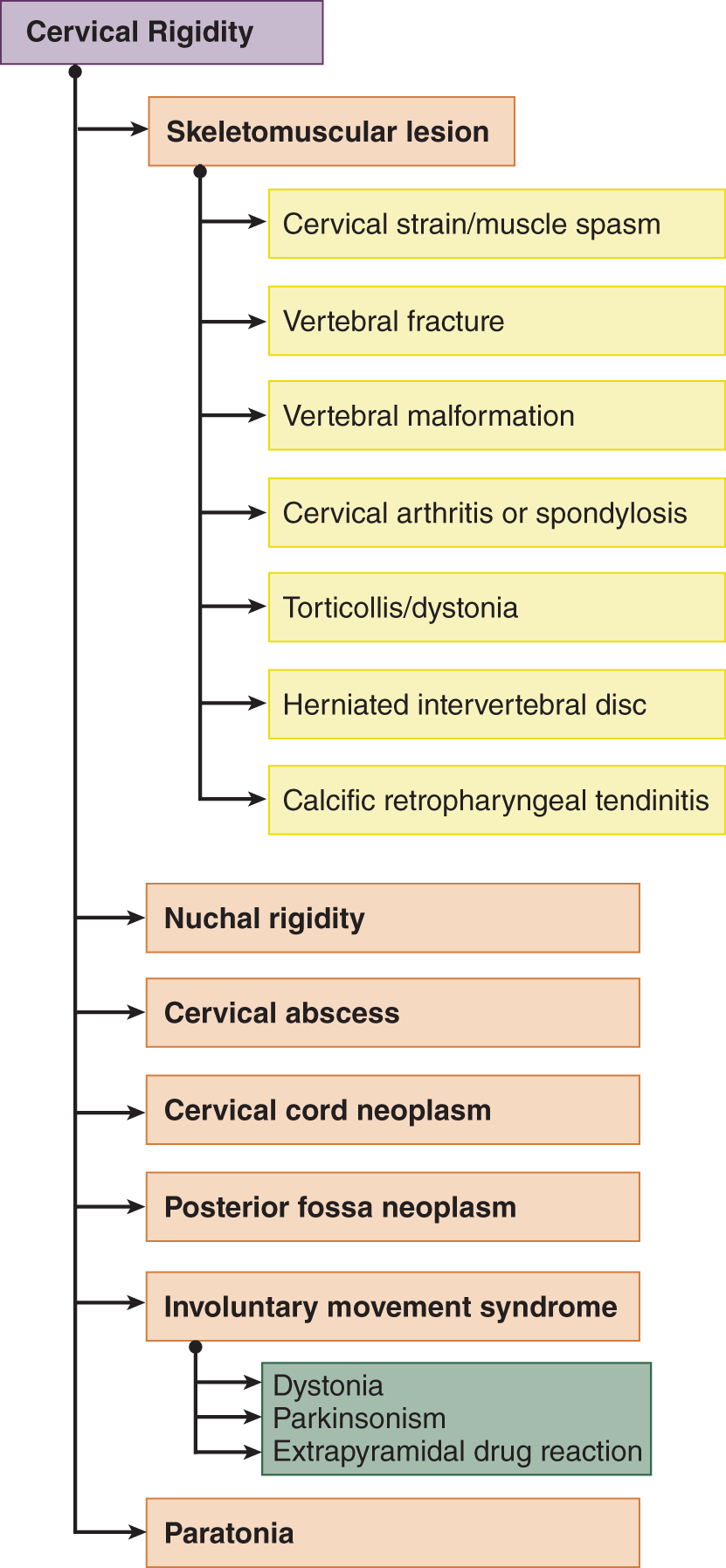
FIGURE 12-26. Dendrogram for causes of cervical rigidity (reference).
I. Neck manipulation and spinal cord injury (Fig. 12-27)
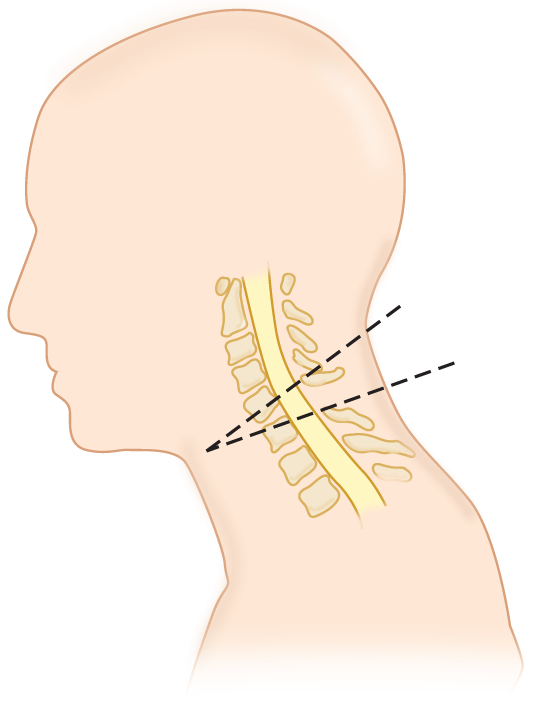
FIGURE 12-27. Fracture and dislocation of C5 on C6. The displaced vertebrae have impinged on the contused and swollen cord.
1. Contraindications to neck manipulation: Study Fig. 12-27, which shows a fracture dislocation of the cervical vertebrae but can represent any mass lesion, such as a tumor or herniation of an intervertebral disc, that compresses the cervical cord.
a. Head flexion would place tension on the swollen spinal cord. The cord will impinge on any intruding mass. For this reason, do not test for nuchal rigidity in a Pt suspected of a cervical cord lesion. Always suspect that any Pt unconscious from trauma has a cervical fracture and obtain cervical spine films (Trunkey, 1991; Gisbert et al, 1989). Be cautious about any neck manipulation in any comatose Pt, particularly a neonate, because the pain-protective reflexes may not be active.
b. In addition to cervical cord injury, neck manipulation may compress the vertebral artery (Fig. 12-34) and cause vertebrobasilar stroke (Garg et al, 1993, Williams and Biller, 2003; Biller et al, 2014) or cause spinal cord ischemia (Linssen et al, 1990). This complication may follow chiropractic manipulation.
2. Positioning of a Pt with known or suspected cervical cord injury: Your natural tendencies urge you to place an injured Pt supine with the head flexed on a pillow, exactly the wrong position. In a suspected cervical cord injury, splint and transport the Pt with the neck in a position to reduce tension on the cord—in other words, in a neutral or slightly _________
3. Lhermitte sign after neck flexion: Neck flexion in the alert Pt may cause a shock-like sensation extending from the neck to the feet. Coughing, sneezing, or straining may elicit the same sensation. The explanation is mechanical stretch of the cord in the presence of a cervical cord lesion causing irritation of the sensory pathways. It is common in multiple sclerosis (Gutrecht, 1989).
J. Review of nuchal rigidity
1. In nuchal rigidity, the neck moves freely in all directions except for _________
2. State the maneuvers to show that a Pt with resistance to neck flexion has nuchal, and only nuchal, rigidity.
_________
_________
3. Then, and only then, is it a reliable sign of an irritative process in the _________
4. If the head of the Pt with decreased consciousness resists movement in all directions, and the extremities do likewise, then the Pt has the generalized hypertonia called _________
K. Confirmatory signs of meningeal irritation: Brudzinski and Kernig signs
1. Brudzinski sign: When testing for nuchal rigidity, watch for adduction and flexion of the legs (Brudzinski sign) as you attempt to flex the head (Wartenberg, 1950). Why would the Pt’s knees flex? _________
2. Leg-raising tests: With the Pt supine, do the bent-knee and straight-knee leg-raising tests of Kernig and Lasègue (Fig. 10-7). Meningeal irritation causes the Pt to resist leg movement in both cases.
L. Absence of meningeal irritation signs in the presence of meningeal irritation
The usual meningeal irritation signs may fail to occur in five circumstances: infancy, senility, coma, with peripheral neuromuscular paralysis, and after acute interruption of the pyramidal tracts. The proof of inflammation or subarachnoid hemorrhage in these Pts requires an LP (Chapter 13). In addition, meningeal leg signs may fail to appear on the side of a hemiplegia. Unilateral absence may aid in identifying the hemiplegic side (Thorner, 1948).
M. Opisthotonos, a driven posture
1. First of all, learn to pronounce the word correctly: ah-piss-THAH-tonos, not oh-PISS-tho-tonos.
2. Definition: Opisthotonos (or Opisthotonus) means a bowed-backward, or hyperextended, position resembling a “wrestler’s bridge” (Fig. 12-28).
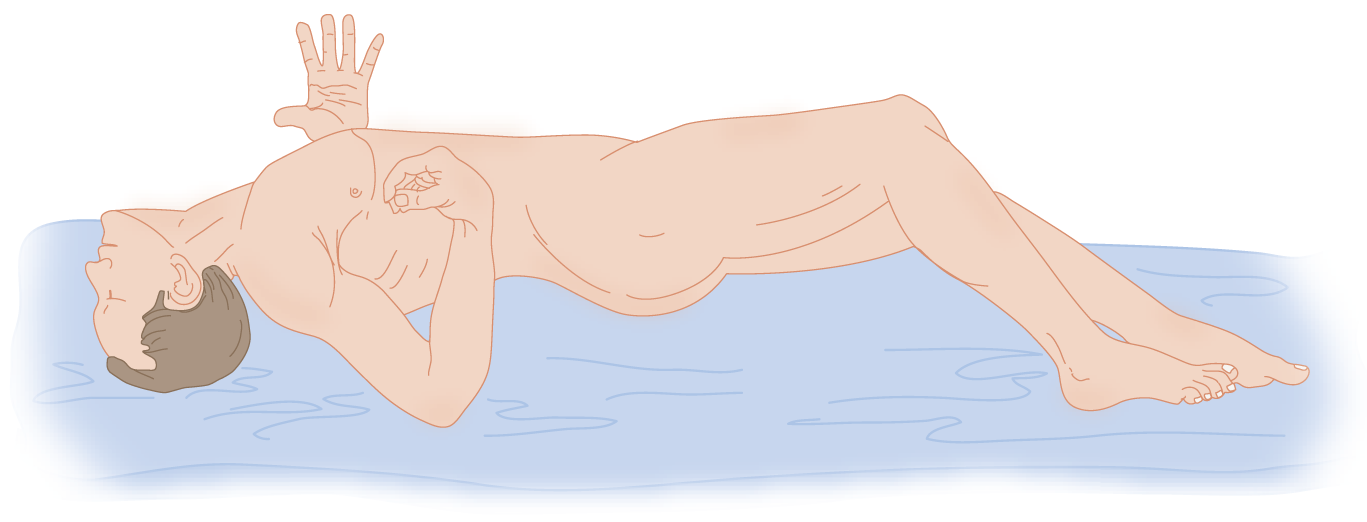
FIGURE 12-28. Severe opisthotonos. (Adapted with permission from Dorland WAN: Dorland’s Illustrated Medical Dictionary. 25th ed. Philadelphia, PA: W.B. Saunders; 1974.)
3. Causes: Opisthotonos results from overcontraction of the immensely powerful extensor muscles of the spine. It occurs with meningeal irritation, decerebrate rigidity, tetanus, and strychnine intoxication, hence, from several different pathophysiologic mechanisms. It may also occur with exposure to centrally acting dopamine blocking agents, and in somatic symptom disorders and schizophrenia (Caroff et al, 2011) (Fig. 12-29).
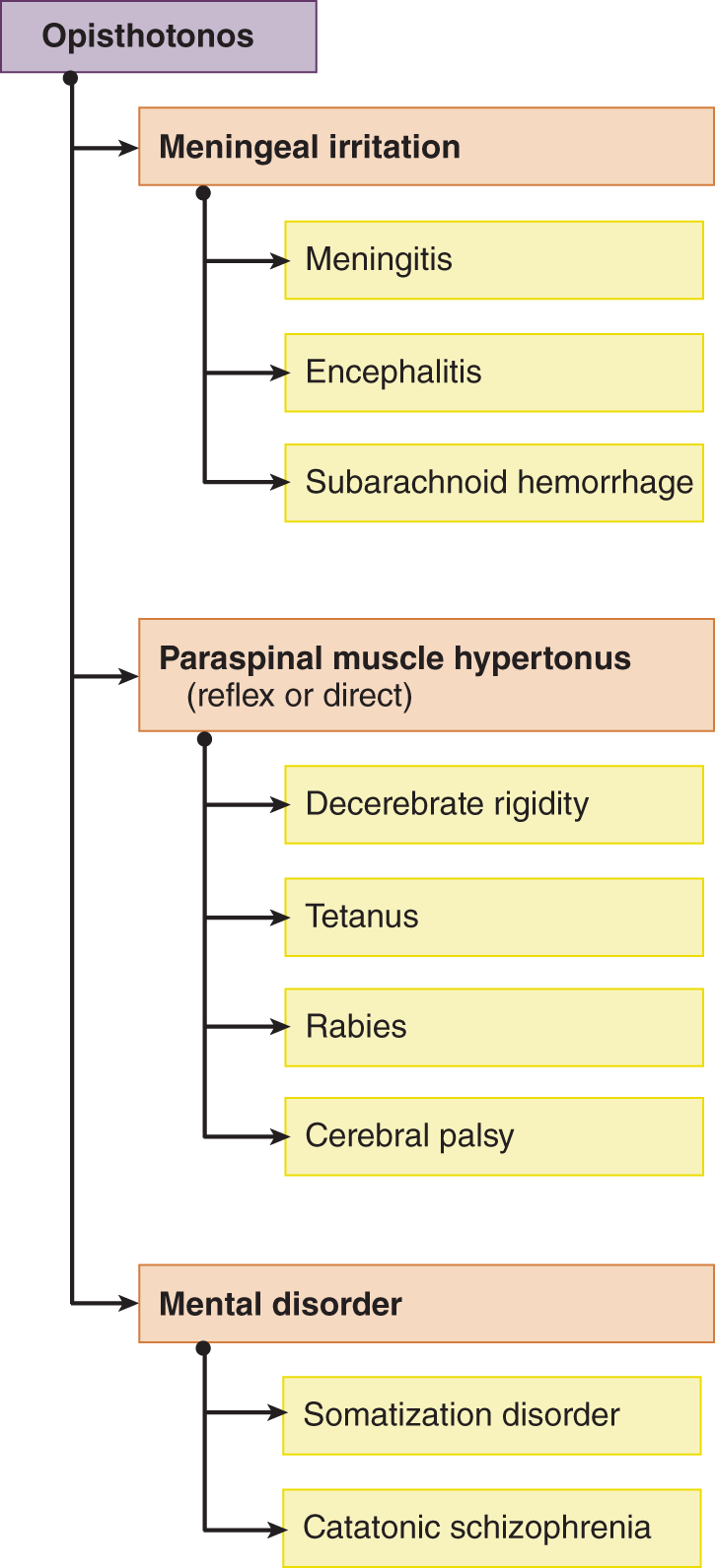
FIGURE 12-29. Dendrogram for causes of opisthotonus (reference).
a. In meningeal irritation, opisthotonos results from spasm of the powerful extensor muscles that splint the neck and back against flexion, which causes pain. We can regard it as a pain-protective reflex posture in these cases.
b. In decerebrate rigidity, the opisthotonic posture results from the hypertonus of the quadrupedal antigravity muscles driven by the vestibular system after an anatomic midbrain lesion or a metabolic lesion that has inactivated or disconnected the cerebrum from the brainstem.
c. In tetanus and strychnine intoxication, MSR and LMN excitability increase tremendously. All skeletal muscles become extremely hypertonic. Tetanus toxin, in addition to its central excitatory effects, has a contractile action on muscles, causing them to remain cramped even after transection of their nerves (Dastur et al, 1977). When all muscles contract maximally, the strongest muscles dictate the posture by overpowering their weaker antagonists. However, in tetanus in particular, local factors based on variability in the excitability of the LMN pools and the direct cramping action of the toxin may alter the strongest muscle law. Thus, as shown in Fig. 12-28, the arm flexors may overcome their stronger antagonists, the extensors. To appreciate what the opisthotonic Pt suffers, lie down supine, extend your neck and legs into the wrestler’s bridge position, clench your teeth (lock-jaw), and contract every muscle in your body as hard as you can for 1 minute.
d. Additional causes of opisthotonos include: seizures, Krabbe disease, glutaric aciduria and organic acidemias, stiff-person syndrome, Chiari malformation, severe electrolyte imbalance, growth hormone deficiency, etc.
4. In summary, opisthotonos results from at least three different pathogenic mechanisms.
a. In decerebrate rigidity:
_________
b. In meningeal irritation:
_________
c. In strychnine or tetanus intoxication:
_________
BIBLIOGRAPHY · Neurologic Examination of the Unconscious Patient
Emery JL. Kinking of the medulla in children with acute cerebral oedema and hydrocephalus and its relationship to the dentate ligaments. J Neurol Neurosurg Psychiatry. 1967;30:267–275.
Fisher CM. The neurological examination of the comatose patient. Acta Neurol Scand. 1969;45 (Suppl 36):1–56.
Jacks AS, Miller NR. Spontaneous retinal venous pulsation: aetiology and significance. J Neurol Neurosur Psychiatry. 2003;74:7–9.
Keane JR. Spastic eyelids: failure of levator inhibition in unconscious states. Arch Neurol. 1975;32:695–698.
Levin BE. The clinical significance of spontaneous pulsations of the retinal vein. Arch Neurol. 1978;35:37–40.
Plum F, Posner J. The Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.
Posner JB, Saper CB, Schiff ND, et al. Plum and Posner’s Diagnosis of Stupor and Coma. 4th ed. New York, NY: Oxford Univ. Press; 2007.
Rizzo MA, Fisher M, Lock JP. Hypermagnesemic pseudocoma. Arch Int Med. 1993;153(9):1130–1132.
Wijdicks EFM, Bamlet WR, Maramattom BV, et al. Validation of a new coma scale: the FOUR score. Ann Neurol. 2005;58:585–593.
Meningeal Irritation Signs: Nuchal Rigidity and Opisthotonos
Anastasopoulous D, Maurer C, Nasiosti G, Mergner T. Neck rigidity in Parkinson’s disease patients is related to incomplete expression of reflexive head stabilization. Exp Neurol. 2009;217(2):336–346.
Biller J, Sacco RL, Albuquerque FC, et al. On behalf of the American Heart Association Stroke Council. Cervical arterial dissections and association with cervical manipulative therapy A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014;45(10):3155–3174. Published online before print August 7, 2014, doi: 10.1161/Stroke 2014;45(10):3155–3174.
Breig A. Adverse mechanical tension in the central nervous system: an analysis of cause and effect. New York, NY: Wiley; 1978.
Caroff SN, Hurford I, Lybrand J, Campbell EC. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011 Feb;29(1):127–148.
Dastur FD, Shahani WR, Dastoor DH, et al. Cephalic tetanus: demonstration of a dual lesion. J Neurol Neurosurg Psychiatry. 1977;40:782–786.
Emery J. Kinking of the medulla in children with acute cerebral oedema and hydro-cephalus and its relationship to the dentate ligaments. J Neurol Neurosurg Psychiatry. 1967;30:267–275.
Garg B, Ottinger CJ, Smith RR, et al. Strokes in children due to vertebral artery trauma. Neurology. 1993;43:2555–2558.
Gisbert VL, Hollerman JJ, New AL. Incidence and diagnosis of C7-T1 fractures and subluxations in multiple trauma patients: evaluation of the advanced trauma life support guidelines. Surgery. 1989;106:702–708.
Goel VK, Weinstein JN. Biomechanics of the spine: clinical and surgical perspective. Boca Raton, FL: CRC Press; 1989.
Gutrecht JA. Lehrmitte’s sign: from observation to eponym. Arch Neurol. 1989;46:557–558.
Hunderfund ANL, Robertston CE, Bell ML, et al. Calcific retropharyngeal tendinitis: unusual cause of neck pain with nuchal rigidity. Neurology. 2008;71(10):778.
Linssen WHJP, Praamstra P, Fons JM, et al. Vascular insufficiency of the cervical cord due to hyperextension of the spine. Pediatr Neurol. 1990;6:123–125.
O’Connell J. The clinical signs of meningeal irritation. Brain. 1946;69:9–21.
Thorner J. Modification of meningeal signs by concomitant hemiparesis. Arch Neurol Psychiatry. 1948;59:485–495.
Trunkey D. Initial treatment of patients with extensive trauma. N Engl J Med. 1991;324:1259–1263.
Wartenberg R. The signs of Brudzinski and Kernig. J Pediatr. 1950;37:679–684.
Williams LS, Biller J. Vertebrobasilar dissection and cervical spine manipulation: a complex pain in the neck. Neurology. 2003;60(9):1048–1049.
V. SENSORY TESTING OF UNCONSCIOUS PATIENTS
A. First principles
1. Necessity for effector integrity: Bedside testing of sensation in the unconscious Pt requires eliciting motor responses. Three conditions may reduce or block the motor response: LMN paralysis, UMN paralysis with cerebral shock, and simply the depth of coma. If no response occurs, then the Ex does not know whether the afferent or efferent limb of the arc has failed. To bypass the necessity for intact effectors, the Ex can assess the integrity of sensory pathways by electronically recording evoked responses to somatosensory, visual, and auditory stimuli (Chapter 13).
2. Follow a rostrocaudal routine: As with an alert Pt, the Ex adopts a rostrocaudal routine to test the sensory systems. Start with the rostralmost CrNs and proceed caudally over the body. Skip CrN I.
B. Testing the pupillary light reflex and cranial nerves II and III
1. Test pupillary reaction to light with a flashlight. If the pupils do not seem to react, darken the room completely and then suddenly turn on the overhead light while watching the pupils through a magnifying glass.
2. Measure and record the size of the pupils in millimeters. Use an actual scale of dots (Fig. 12-30).
FIGURE 12-30. Scale in millimeters for measuring pupillary size.
3. Many factors can alter pupillary size (Table 12-6). Always consider the possibility that the Pt has taken a drug that can alter pupillary size.
TABLE 12-6 • Pupillary Findings in the Unconscious Patient
Size and reactions |
Toxic-metabolic agents |
Anatomic lesions |
Widely dilated, nonreactive |
Strong cholinergic blocker or sympathomimetic drugs |
Midbrain failure or bilateral CrN III lesions |
Widely dilated but reactive |
Mild anticholinergics in some OTC sleeping and cold medications; adrenergic vasopressors |
May mean early midbrain failure |
Midposition or mildly dilated (4–6 mm) and sluggish or nonreactive |
Glutethimide (Doriden) |
May mean both sympathetic and parasympathetic paralysis: Adie Syndrome |
Pupilloconstriction |
Sleep, anoxia, uremia; glaucoma drugs or other parasympathomimetics |
Interruption of descending sympathetic pathway |
Pinpoint but reactive if bright light used |
Opiates |
Massive pontine lesion, often a hemorrhage, that disrupts descending sympathetic pathways |
ABBREVIATIONS: CrN = cranial nerve; OTC = over the counter. |
||
4. The pupillary light reflex involves CrNs II (afferent) and III (efferent) simultaneously. In Fig. 12-31, draw the pupilloconstrictor reflex pathway. Start, as usual, at the receptor and trace through the pathway.
FIGURE 12-31. Blank to draw the pathway of the pupilloconstrictor reflex. Start with a retinal neuron. Check your drawing against Fig. 4-30, page 151.
C. Technique for testing sensory function of cranial nerve V
1. Corneal reflex (V afferent, VII efferent): Test with a wisp of cotton, as in the conscious Pt.
2. Supraorbital compression test for reflex of CrNs V to VIII
a. Press your thumbnail strongly into the Pt’s eyebrow over the superciliary notch, the exit site of the ophthalmic division of CrN V. Locate your own notch by pressing the ball of your thumb from medial to lateral along your eyebrow. Then press your fingernail in until you feel pain.
b. With an intact pons and CrNs V and VII, the unconscious Pt responds with an ipsilateral facial grimace with eyelid closure. Hence, the Ex can test each half of the face separately. In the comatose Pt with acute hemiplegia, the grimace would be absent or weaker on the hemiplegic/ nonhemiplegic side. Explain. _________
_________hemiplegic) (After acute UMN lesions, the excitability of reflexes is temporarily reduced during the phase known as cerebral shock.)
c. If the Ex suddenly and unexpectedly flicks the closed eyelashes of the Pt in feigned coma, the Pt often shows an exaggerated response with grimacing and head retraction. This is practically never seen in organic coma (Wiggs, 1973).
D. Testing hearing: the auditory division of cranial nerve VIII (auditopalpebral reflex)
Test by a sudden sound, such as a clap or clanging metal pans together, and observe the Pt for a startle response.
E. Testing the vestibular division of cranial nerve VIII
Two tests, caloric irrigation and the counter-rolling test, elicit a vestibulo-ocular reflex that causes conjugate deviation of the eyes. The receptors for the tests are essentially the cupulae of the semicircular canals, but the cristae and neck proprioceptors may add somewhat to the response (Buettner and Zee, 1989). The vestibular nuclei relay the vestibular impulses through the MLF to activate CrNs VI and III.
1. The counter-rolling test for the vestibulo-ocular reflex (doll’s eye test or oculocephalic reflex)
a. Principle of the test: If you have an old-fashioned doll, hold it up in front of you. If you tilt the doll’s head down, the eyes will turn up to remain looking at you in the eye. If you tilt the doll’s head up, the eyes rotate down to remain looking at you in the eye. Counterweights behind the eyes cause this counter-rolling action, which essentially keeps eyes on the visual target when the head moves.
b. Technique for the counter-rolling test: Hold the Pt’s head in a neutral position with your hands and briskly rotate it to the right and left and up and down, and stop it in each rotated position. For example, rotate the head to the right and stop it in that position. During the initial rotation to the right, the eyes counter-roll against the direction of head turning, thus turning to the left relative to the head, but actually the eyes simply maintain their original position. As the Ex holds the head in the rotated position, the eyes quickly turn and align straight ahead for the new head position. Failure of the eyes to counter-roll indicates failure of the vestibulo-ocular reflex.
2. Review of caloric irrigation
a. Indication: If the counter-rolling test fails, do caloric irrigation. Caloric irrigation provides a more powerful stimulus than head turning, but the counter-rolling action added to caloric irrigation is more effective than either alone (Fisher, 1969).
b. Precaution: What precaution must you take before injecting water into the external auditory meatus? _________
c. Technique for caloric irrigation: Review the technique for caloric irrigation from Chapter 9. Give details of position, equipment, and water temperature. _________
_________
d. Normal response to cold caloric irrigation: The Pt shows nystagmus featured by slow deviation of the eye ipsilaterally and quick, saccadic jerks that restore the eyes to the primary position. As the coma deepens, the frontopontine pathway fails and the saccades disappear, but the vestibular deviation remains. In yet deeper coma, the deviation lessens until no response is obtained at all, even though the pathways remain anatomically intact.
e. If the Pt shows no response, you have to “think circuitry” to locate the reason for the failure:
i. Start with the nature of the stimulus. The stimulus may have been inadequate. Try colder water and irrigate longer.
ii. The Pt’s labyrinths are inexcitable or CrNs VIII are interrupted.
iii. The vestibular nuclei or MLF are interrupted by an extensive tegmental lesion.
iv. The peripheral nerves to the ocular muscles are interrupted, or neuromuscular transmission has failed.
v. The Pt may be in a stage of profound coma or may be brain dead. Obviously, any response provides more useful information than no response.
f. Significance: Normal pupillary reactions to light and full normal conjugate counter-rolling of the eyes in response to the vestibulo-ocular stimuli virtually exclude an anatomic lesion of the pontomesencephalic tegmentum as the cause for the unconsciousness. The Pt has a supratentorial lesion or metabolic-toxic coma. Fixed eyeballs, and thus no vestibulo-ocular reflex, with normally reactive pupils, suggest depressant drugs, generally barbiturates.
3. Summarize three methods used to investigate the comatose Pt for the integrity of the MLF in its course through the tegmental core of the brainstem. _________
4. Which method should you choose to test the MLF in a Pt unconscious from head trauma? caloric irrigation/ counter-rolling test. Explain.
_________
_________caloric irrigation) (Do not manipulate the neck in a Pt with suspected head trauma because of the possibility of a cervical fracture.)
5. An alternative to caloric irrigation is galvanic stimulation of the labyrinth (Toglia et al, 1981).
F. Cranial nerves IX and X
Spontaneous swallowing or groaning constitutes a sufficient test. Actually eliciting the gag reflex may induce vomiting and aspiration of stomach contents into the lungs. However, see the brain-death protocol (BDP), page 504.
G. Techniques for eliciting responses to noxious stimuli: cold and pain
1. To test the somatic pathways in unconscious Pts, use stimuli that a conscious Pt would perceive as noxious or painful. First, make a decision whether to use cold or pain. Cold is often the more appropriate stimulus for infants and children (especially when the parents are looking on) and for partly conscious Pts.
2. Technique to test for response to cold: Apply an ice cube to the Pt’s face, hands, and feet. If there is no response, apply it to armpits, abdomen, or inner thighs (actions that will get your attention quickly).
3. Techniques to test for pain responses:
a. Mandibular test for pain response: Place your fingers on the temporomandibular joints and press (Wijdicks, 2001) or place your fingers behind the ramus of the mandible and compress the tips against the bone, as if to pull the mandible forward. This provides a very strong pain stimulus. Try it on yourself and a partner.
b. Knuckle rub tests for pain response: Make a fist and press or rub your knuckles strongly against the Pt’s sternum. Observe for an arousal response with eye opening or decerebrate posturing.
c. Nail compression tests for pain: Test for localized pain responses by stimulating each extremity separately. Pinch the Pt’s fingernails or toenails hard, or press them with the rubber tip of a pencil. To ensure a painful stimulus, the Ex must apply the pinch correctly. Grasp your left thumbnail between the tip of the right thumbnail and index finger and pinch it hard. You may or may not feel much pain. Next grasp your left thumbnail by placing the center of the tip of the right thumb over the margin of the left thumbnail where it attaches to the flesh, and this time squeeze and torque, or twist, the left nail, as though trying to screw on a bottle cap. Try the technique with other fingers. With practice you can learn to torque only the fingernail, not the entire digit, which may injure the Pt’s finger joints.
d. Skin pinch test for pain response: As an alternative pain stimulus, try pinching the Pt’s skin between your thumb tip and index finger. In this era of acquired immunodeficiency syndrome and infectious hepatitis, avoid pinpricks. They often leave a trail of dots of blood. Also do not use your fingernails, except to compress the supraorbital nerve, where the eyebrow provides protection, or you may leave visible wounds. Try the various ways of testing facial and body pain on yourself to learn the proper strength of stimulus to use.
4. Do not overdo pain testing in the Pt suspected of feigned coma. The Pt may have a steely resolve not to respond. Sudden, unexpected stimuli, such as flicking a closed eyelid, are more likely to elicit a response than extremely strong ones (Wiggs, 1973).
5. Record the result of pain testing in the Glasgow Coma Scale. In general, the arm responses are more informative than the leg responses because a purely spinal reflex can cause flexion of the leg in a Pt with a transected cord or in the brain-dead Pt whose spinal cord remains intact.
6. Review the mandibular, sternal rub, nail, and skin pinch tests for responses to pain in the comatose Pt.
BIBLIOGRAPHY · Sensory Testing of Unconscious Patients
Buettner UW, Zee DS. Vestibular testing in comatose patients. Arch Neurol. 1989;46:561–563.
Fisher CM. The neurological examination of the comatose patient. Acta Neurol Scand. 1969;45 (Suppl 36):1–56.
Toglia JU, Adam RU, Steward G. Galvanic vestibular tests in the assessment of coma and brain death. Ann Neurol. 1981;9:294–295.
Wijdicks EF. The Diagnosis of Brain Death. N Eng J Med. 2001;344:1215–1221.
Wiggs JW. Detection of feigned coma. N Engl J Med. 1973;289:379.
VI. FACTORS THAT COMPOUND AND COMPLICATE THE NEUROLOGIC EXAMINATION OF THE UNCONSCIOUS PATIENT
A. The muscle stretch reflexes and toe signs
The MSRs and toe signs that prove so valuable in the alert Pt take a secondary role in coma. You may find decreased, equal, or increased MSRs or no MSRs depending on cerebral shock and the depth of the coma. Even when the MSRs are unequal, you may not know whether one side is pathologically depressed or the other pathologically active. Depression of consciousness for any reason, even deep sleep in some individuals, may cause bilateral extensor toe signs. Thus, in coma, neither the presence nor the absence of the typical UMN signs have the significance that they do in the alert Pt.
B. Preexisting neurologic signs
Preexisting conditions may confuse the diagnosis. Is a given finding preexistent or new? The apparent unconsciousness may be the result of petit mal or psychomotor status epilepticus; the failure to react to sound may represent preexisting deafness; the immobile eye with the nonreactive pupil may be a glass eye; the Pt may have congenital ptosis or anisocoria; the pupils may react sluggishly; the MSRs may be absent because of Adie syndrome (Table 4-5); the hemiplegia may be a postictal paralysis after a focal seizure (Todd paralysis); or the Pt may have had a preexisting hemiplegia or other neurologic signs before the coma. Mild diffuse atrophy, brisk MSRs, and spasticity, particularly of the fingers, suggest a preexisting hemiplegia. Photographs and family members can document preexisting illness.
C. How to address the unconscious patient: the law of respect
Because of the difficulties in judging the level of consciousness, the Ex must never, never, make flippant or pejorative remarks at the bedside of a presumably unconscious or anesthetized Pt or discuss prognosis or management controversies. Anesthetized or unconscious Pts often hear and remember more than expected (Baier and Schomaker, 1990; Cheek, 1960). Always assume that the Pt hears and understands everything said at the bedside. The physician, ward personnel, and family should address the Pt by name and talk to the Pt as if he were conscious (LaPuma et al, 1988). Although such stimulation may or may not speed recovery (Pierce et al, 1990), addressing unconscious Pts by name and avoiding thoughtless comments cause no harm and does affirm the Pt’s status as a human being rather than as just a carcass.
BIBLIOGRAPHY · How to Address the Unconscious Patient
Baier S, Schomaker MS. Bed Number Ten: A Patient’s View of Long Term Care. Boca Raton, FL: CRC Press; 1990.
Cheek DB. What does the surgically anesthetized patient hear? Rocky Mtn Med J. 1960;57:49–53.
LaPuma J, Schiedermayer DL, Gulyas AE, et al. Talking to comatose patients. Arch Neurol. 1988;45:20–22.
Pierce JP, Lyle DM, Quine S, et al. The effectiveness of coma arousal intervention. Brain Inj. 1990;4:191–197.
VII. THE NEUROLOGIC EXAMINATION IN THE LOCKED-IN SYNDROME (DE-EFFERENTED STATE)
You survive, but you survive with what is so aptly known as “locked-in syndrome”. Paralyzed from head to toe, the patient, his mind intact, is imprisoned inside his own body, unable to speak or move. In my case, blinking my left eyelid is my only means of communication.
A. Clinical features
1. Bilateral extensive lesions—infarction, trauma, demyelination, or neoplasm—may destroy both pyramidal tracts in the basis pontis or midbrain (Chia, 1991; Feldman, 1971; Reznick, 1983; Turazzi and Bricolo, 1977). The Pt suffers complete bilateral hemiplegia, anarthria, aphagia, and incontinence. The Pt retains full consciousness and sensation because the lesion spares the tegmentum and its contained reticular formation and lemnisci. Breathing continues automatically, but the only volitional movements remaining are vertical eye movements and sometimes blinking. The pathways for these actions enter the pretectum and tectum rostral to the lesion. Usually the lesion interrupts the pathways to the conjugate lateral gaze center in the pons, causing paralysis of volitional horizontal eye movements (Nordgren et al, 1971; Fig. 2-30). The Pt, although conscious, may display decerebrate posturing to stimuli (Feldman, 1971; Nordgren, 1971).
2. The Ex can communicate with the Pt and establish consciousness by the only volitional effector mechanism remaining, the eye movements, by using upward movements for yes and downward for no. Otherwise, the Pt’s consciousness, deprived of any efferent pathways to other effectors, remains “locked-in.” In rare instances when the lesion destroys the pyramidal tracts at the midbrain level, the Pt may also retain volitional blinking and horizontal eye movements (Chia, 1991).
3. If unrecognized by the Ex, the locked-in state condemns the Pt to the cruelest of existences. Perfectly conscious and in possession of all senses and faculties, the Pt cannot avoid even the most trifling annoyances. The Pt cannot adjust a heel to relieve pressure, scratch an itch, flick a fly from the face, or voluntarily swallow to relieve a flooded pharynx. Although thought and emotion remain as powerful as ever, the Pt cannot utter the faintest whisper or write a single letter. Even the total helplessness is not the worst of it, if others make unintended but dehumanizing remarks at the bedside, as if the Pt were a non-person. Moreover, because a Pt may pass through the locked-in state during recovery from coma, the Ex must always anticipate the return of sentiency and monitor for it daily by meticulous NEs.
BIBLIOGRAPHY · The Locked-in Syndrome
Bauby JD. The Diving Bell and the Butterfly. Random House; New York, New York, 1997.
Chia L-G. Locked-in syndrome with bilateral ventral midbrain infarcts. Neurology. 1991;41:445–446.
Feldman MH. Physiological observations in a chronic case of “locked-in” syndrome. Neurology. 1971;21:459–478.
Nordgren RE, Markesbery WR, Fukuda K, et al. Seven cases of cerebromedullospinal disconnection: the “locked-in” syndrome. Neurology. 1971;21:1140–1148.
Reznick M. Neuropathology in seven cases of locked-in syndrome. J Neurol Sci. 1983;60:67–78.
Turazzi S, Bricolo A. Acute pontine syndromes following head injury. Lancet. 1977 Jul 9;2(8028):62–64.
VIII. THE NEUROLOGIC EXAMINATION IN THE PERSISTENT VEGETATIVE STATE
A. Definition
Jennett and Plum (1972) defined the persistent vegetative state (PVS) as a persistent state after coma, characterized by eye opening and “a return of wakefulness accompanied by an apparent total lack of cognitive function.” In essence, the Pt, appears awake but remains unaware of self and environment. Awareness is abolished but arousal from sleep persists, thus implying the separation of these two functions.
1. Persistent emphasizes that the PVS has already lasted for some time, 4 weeks at a minimum, and implies that the Pt may remain that way for an extended period.
2. Vegetative emphasizes the assumption that the Pt, devoid of mind or volitional actions, retains only autonomic, vegetative, or reflex functions (Munsat et al, 1989).
3. Some states such as the akinetic mutism of Hugh Cairns (1941, 1952), the decorticate or apallic state of Kretchsmer (Dalle et al, 1977), and the coma vigile of the French resemble, or are synonymous with, the PVS, but sometimes coma vigile is identified with the locked-in syndrome (Victor and Ropper, 2001).
B. Neuropathology of the persistent vegetative state
1. The most common causes of the PVS are severe hypoxia, head trauma, or massive infarction, resulting in permanent bilateral diffuse damage, usually at multiple levels from the rostral brainstem through diencephalon, basal ganglia, deep white matter, and cerebral cortex (Adams et al, 2000; Jennett, 2002; Kinney and Samuels, 1994). After such a catastrophe, the stage of eyes-closed unconsciousness usually lasts no more than a month. Then the Pt will enter the stage of eyes-open unconsciousness, the so-called PVS.
2. Patients with progressive cerebral degenerative diseases may also end in a vegetative state, and some infants with severe brain malformations meet the criteria (Multi-Society Task Force, 1994, Part 1; 1994, Part 2). Estimates place the number of PVS Pts in the United States as between 10,000 and 25,000 adults and 4000 and 10,000 children (Multi-Society Task Force, 1994, Part 1; 1994, Part 2).
C. Clinical features of the persistent vegetative state (Jennett, 2002)
1. The Pts cannot sit, stand, walk, or talk. The Pts remain mute and completely devoid of any verbal or nonverbal communication or of any purposeful or adaptive behavior that offers operational proof of consciousness or volition.
2. PVS Pts are akinetic or hypokinetic. Although the Pt may display more or less severe paralysis, spasticity, rigidity, or paratonia and sometimes hypotonia, the akinesia is out of proportion to and not directly caused by a UMN syndrome. The Pt cannot do any self-help tasks such as dressing or feeding.
3. PVS Pts exhibit apparent sleep and wake cycles. They open their eyes during the day or sometimes in response to putatively painful stimuli.
4. PVS Pts may blink, move their eyes spontaneously, and may fixate and pursue to some degree. They do not actively fixate and selectively move the eyes from target to target, which would imply consciousness. Vestibulo-ocular reflexes generally remain.
5. PVS Pts flex their limbs in response to pain but show no purposive or adaptive avoidance. They fail to orient actively or adaptively to light, sound, or touch but may startle reflexively in response to these stimuli.
6. PVS Pts retain many autonomic and somatic reflexes. They are incontinent for urine and feces but generally support their own blood pressure and breathe automatically.
7. PVS Pts show many of the primitive reflexes reviewed in Table 11-6: grasping, sucking, automatic yawning, and chewing, but not purposeful chewing and swallowing. They may show cyclic automatic chewing movements unrelated to feeding. Feeding is by tube or gastrostomy. Some Pts display the spontaneous crying or laughing of pseudobulbar palsy (Higashi et al, 1981).
D. The minimally conscious state
Some Pts fall somewhere beyond a true PVS but are far from fully conscious (Table 12-8; Giacino et al, 2002). They show inconsistent but at least some discernible behavioral signs suggestive of consciousness. This diagnostic category remains in need of further study before full acceptance (Bernat, 2002; Coleman, 2002).
E. Prognosis for survival and recovery from the persistent vegetative state
1. Survival time in the PVS has increased with improvement in overall management (Strauss et al, 1999). Children have a greater potential for survival than adults (Kriel et al, 1993). Operational proof of consciousness has returned to some Pts who have been in the PVS for as long as 18 to 36 months (Arts et al, 1985; Higashi et al, 1981; Rosenberg, 1977) and anecdotally after more than a decade. Seen in this light, the PVS may represent a phase, after coma, preceding the return of consciousness. For these reasons, the Ex must periodically repeat the NE in all Pts with altered consciousness and must not hastily dismiss the Pt as a “vegetable” or too quickly terminate support. The critical question is how long do we wait on a Pt in the PVS to recover consciousness before giving up? As a final confounding factor, even though a person in the PVS or minimally conscious state had previously given directives about life support, further reflection may change the Pt’s mind. Patients who wanted to die under one circumstance may later change their minds and want to live (Patterson, 1993).
2. The Pt in the “minimally conscious state” further complicates matters (Wijdicks, 2006). The burden of decision challenges the epistemology of the NE, but we have had no better way. Magnetic resonance imaging spectroscopy of the thalamus, a crucial neuronal station in the neuroanatomy of consciousness, shows differences in Pts who recovered from the PVA as contrasted to those who remained in that state (Uzan et al, 2003). In any event the burden does not rest on the Pt to prove “worthiness,” consciousness, and the existence of personhood but on the neurologic evaluation to refute these attributes. If all this uncertainty about our ability—yours and mine—to judge consciousness worries you, you have the right attitude. Conversely, if the PVS exemplifies the failure of the NE to provide an absolute solution, the brain-death protocol (next section) fully demonstrates the validity and power of the NE. Most neurologists agree that the diagnosis of brain death is straightforward, reliable, and uncontroversial.
BIBLIOGRAPHY · The Neurologic Examination in the Persistent Vegetative State
Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after an acute brain insult. Brain. 2000;123:1327–1338.
Arts W, van Dongen H, Van Hof-van Duin J, et al. Unexpected improvement after prolonged posttraumatic vegetative state. J Neurol Neurosurg Psychiatry. 1985;48:1300–1303.
Bernat JL. Questions remaining about the minimally conscious state. Neurology. 2002;58:337–338.
Cairns H. Disturbances of consciousness with lesions of the brainstem and diencephalon. Brain. 1952;75:109–146.
Cairns H, Oldfield R, Pennybacker JB, et al. Akinetic mutism with an epidermoid cyst of the third ventricle. Brain. 1941;64:273–290.
Coleman D. The minimally conscious state: definition and diagnostic criteria. Neurology. 2002; 58(3):506–507.
Dalle Ore G, Gerstenbrand F, Lucking CH, et al, eds. The Apallic Syndrome. New York, NY: Springer-Verlag; 1977.
Giacino JT, Ashwal S, Childs N, et al. The minimally conscious state: definition and diagnostic criteria. Neurology. 2002;58:349–353.
Higashi K, Hatano M, Abiko S, et al. Five-year follow-up study of patients with persistent vegetative state. J Neurol Neurosurg Psychiatry. 1981;44:552–554.
Jennett B. The Vegetative State. Medical Facts, Ethical and Legal Dilemmas. New York, NY: Cambridge Univ. Press; 2002.
Kinney HC, Samuels MA. Neuropathology of the persistent vegetative state: a review. J Neuropath Exp Neurol. 1994;53:548–558.
Kriel RL, Krach LE, Jones-Saete C. Outcome of children with prolonged unconsciousness and vegetative states. Pediatr Neurol. 1993;9:362–368.
Multi-Society Task Force. Medical aspects of the persistent vegetative state. Part 1. N Engl J Med. 1994;330:1499–1508.
Multi-Society Task Force. Medical aspects of the persistent vegetative state. Part 2. N Engl J Med. 1994;330:1572–1579.
Munsat TL, Stuart WH, Cranford RE. Guidelines on the vegetative state: commentary on the American Academy of Neurology statement. Neurology. 1989;39:123–124.
Patterson DR, Miller-Perrin C, McCormick TR, et al. When life support is questioned early in the care of patients with cervical level quadriplegia. N Engl J Med. 1993;328:506–509.
Rosenberg GA, Johnson SF, Brenner RP. Recovery of cognition after prolonged vegetative state. Ann Neurol. 1977;2:167–168.
Strauss DJ, Shavelle RM, Ashwal S. Life expectancy and median survival time in the permanent vegetative state. Pediatr Neurol. 1999;21:626–632.
Uzan M, Albayram S, Dashti SGR, et al. Thalamic proton magnetic resonance spectroscopy in vegetative state induced by traumatic brain injury. J Neurol Neurosurg Psychiatry. 2003;74:33–38.
Victor M, Ropper AM. Adams and Victor’s Principles of Neurology. 7th ed. New York, NY: McGraw-Hill; 2001.
Wijdicks EFM. Minimally conscious state vs persistent vegetative state: the case of Terry (Wallis) vs the case of Terry (Shiavo). Mayo Clin Proc. 2006;81(9):1155–1158.
IX. THE NEUROLOGIC EXAMINATION IN THE DIAGNOSIS OF BRAIN DEATH
A. Traditional definition of death
The traditional definition of death reflected the Aristotelian notion that placed consciousness and, therefore, personhood in the heart. Until about 1960, the criterion for death was permanent cessation of the heartbeat and breathing—that is to say, when these functions stopped and did not recommence spontaneously. Now most physicians accept brain death, the irreversible loss of all brain functions, as the end of personhood and the life of that individual (American Academy of Neurology, 1995; Walker, 1985; Wijdicks, 2001a, 2001b, 2002).
B. Changes in the definition of death to brain death
1. Advances in medical knowledge and medical technology have changed the diagnosis of death. The advance in medical knowledge consisted of universal acceptance of the brain as the site of consciousness and of personhood. Transplantation of the heart, without change in personality of the recipient, was the absolute and final proof of the brain as the site of personhood and eliminated the heart.
2. The two advances in medical technology consisted of:
a. Techniques to maintain or restart breathing and the heartbeat when, formerly, they would have irreversibly stopped.
b. Techniques of organ transplantation (first kidney transplant in 1954; first heart transplant in 1967). Harvesting of organs requires prompt, absolute determination of brain death. These developments rendered the traditional definition of death obsolete.
c. The United States now generally accepts death as the irreversible cessation of all brain function (Wijdicks, 2001a, 2001b, 2002). The United Kingdom and some other countries define death as irreversible cessation of all brainstem function.
C. The brain-death protocol and ethical considerations in brain death
1. The brain-death protocol (Table 12-7) outlines a sequence of operations that enable physicians to recognize and certify—in a word, diagnose—that the brain has died and therefore that the person has died. The physician and society have no ethical responsibility or purpose in maintaining a brain-dead body. Brain death justifies ending all therapy and support and, depending on previously expressed Pt preferences, justifies the removal of organs for transplantation. Consensus on the concept of brain death is widespread throughout the world, but differences of opinion exist, especially with the need for confirmatory procedures beyond the clinical examination (Wijdicks, 2002).
TABLE 12-7 • Criteria for the Brain-Death Protocol
A. Primary clinical criteria 1. The Pt has suffered a clinical disaster that could cause brain death and is completely comatose, with no respiration or other behavior that could arise in the brain. 2. The Pt is receiving artificial respiration and is adequately oxygenated. 3. The Pt has a minimum core body temperature of 36.1°C for a child or 32.2°C for a mature individual. 4. The Pt is normotensive (but may require dopamine, etc., for support) and is in as good a metabolic balance as the circumstances allow. 5. The Pt has no significant level of depressant or neuroparalytic (synaptic-blocking) medications. Determine by history, chart review, and toxicology screen. 6. The Pt shows no brainstem functions whatsoever. a. Absolute coma. b. Midposition or dilated pupils, nonreactive to light, and not paralyzed by neuroactive drugs. c. Absolutely no spontaneous or induced eye movements. d. Absolutely no response to V nerve stimulation: no glabellar blink, corneal reflex, or oculocardiac reflex. e. Absolutely no spontaneous or reflex facial movements. f. Absolutely no oropharyngeal responses: no gag reflex, sucking, chewing, snouting, or rooting, and no tongue movements. g. Absolutely no auditopalpebral or vestibulo-ocular reflexes. h. Complete apnea on adequate testing by temporary stoppage of the respirator. 7. A second complete NE done 6-24 h after the first demonstrates absolutely no responses. B. Ancillary diagnostic tests for brain death 1. Isoelectric EEG. 2. Absence of cerebral blood flow by radionuclide or transcranial Doppler ultrasound studies or conventional angiography. |
ABBREVIATIONS: EEG = electroencephalography; NE = neurologic examination; Pt = patient. |
2. The BDP uses exactly the same principles and procedures as those applied to diagnose any other condition, for example, gangrene of the leg or hepatitis. The process of diagnosing brain death and terminating support for a brain-dead body is entirely separate from and has nothing to do with euthanasia. Euthanasia means killing a person who has a living brain. After brain death, no person remains to be killed, and that is what the BDP establishes.
D. Assumptions underlying the brain-death protocol
1. Because personhood and consciousness reside in the brain, the death of the brain is the death of the person, irrespective of the state of the other organs.
2. Competent physicians can reliably—essentially infallibly—distinguish a live brain from a dead brain. The living brain, if at the right temperature and not simply depressed by toxic–metabolic states, displays:
a. Clinically demonstrable functions
b. Recordable electrical activity
c. Blood flow
3. Conversely, a dead brain displays:
a. No clinically demonstrable functions
b. No recordable electrical activity
c. No blood flow
E. Causes for brain death
The Pt must have suffered a known catastrophe that could have caused brain death. Thus the medical history is an integral part of the BDP. The medical catastrophes that commonly cause brain death are hypoxia, irreversible brain edema, head injuries, and necrotizing meningitis or encephalitis. The Pt is always intubated for mechanical ventilation, is catheterized, and has an intravenous or intra-arterial line to monitor and control the blood pressure and metabolism.
F. Initiation of the brain-death protocol
The attending physician who suspects brain death requests a consultation by a specialist in neurology or neurosurgery. The physician does not generally request a BDP until at least 12 to 24 hours after the medical catastrophe that jeopardized the brain, to allow some time to assess any potential for recovery of the brain.
G. Role of the neurologic specialist in the brain-death protocol
1. The neurologist completes a printed BDP that lists the criteria for brain death (Table 12-7). Steps 2 to 5 of Table 12-7 ensure that the brain is not too cold or too depressed by metabolic imbalance or drugs or too poorly oxygenated to display functions if it remains alive. The Ex must exclude neuromuscular-blocking drugs, depressant drugs, or severe Guillain–Barré syndrome (Hassan and Mumford, 1991) that would prevent the motor responses, the behavior relied on to demonstrate brain function. A toxicology screen is recommended.
2. The Ex does the most complete cranial nerve examination possible (Table 12-7, steps 6a to 6g). The pupils in the brain-dead Pt are usually in midposition and absolutely unreactive. Because the endotracheal tube obstructs visibility in the mouth, the Ex inserts a gloved finger along the side of the tongue and palpates it for any movements and then stimulates the palatal arch for a gag reflex. The Ex decides whether to order an EEG, short latency evoked potentials (Facco et al, 1990) or radionuclide studies (Zuckier and Kolano, 2008), transcranial Doppler ultrasound, or conventional angiography (Posner et al, 2007; Wijdicks, 2001a, 2001b, 2002).
H. Apnea testing in the brain-death protocol
1. If the NE, EEG, or flow study fail to demonstrate any brain function, the Ex tests for apnea. Any brain function shown by the NE or the laboratory tests terminates the BDP at that point, obviating the apnea test. The Ex does the apnea that last because it is the only test of the BDP that can cause harm. Hypoxia during apnea testing can cause ventricular fibrillation and cardiac arrest and may add further anoxic insult to any surviving brain tissue (Goudreau, 2000).
2. For apnea testing, like the other parts of the BDP, the body temperature must be at least 36.5°C (97°F), the metabolic state must be stabilized, and systolic blood pressure must be at least 90 mm Hg. Baseline blood gases are drawn. An oxygen cannula is placed at the carina with a flow of 6 L of O2 for 10 minutes. For the apnea test, the Ex turns off the ventilator for 8 to 10 minutes to achieve a partial pressure of carbon dioxide (PCO2) level of 60 mm Hg or more or that raises the baseline PCO2 more than 20 mm Hg, thus providing a maximum stimulus to the pontomedullary respiratory centers (Gutmann and Marino, 1991; Marks and Zisfein, 1990). The PCO2 usually rises 30 mm/min. During the entire process, the Ex checks for any respiratory efforts by placing one hand flat, spanning the lower ribcage and abdomen, and the other on the mouth, jaw, and neck muscles. Palpation provides a more sensitive and certain way to detect slight respiratory movement than sight. The Ex cannot maintain absolute vigilance by sight for many minutes, but can do so by employing touch. Determine blood gases at the end of the apnea period to ensure that the PCO2 has reached a level that will maximally stimulate the Pt’s respiratory center.
3. Depending on circumstances, the Ex repeats the NE and the apnea test in no less than 6 hours, preferably 12 to 24 hours.
4. Given that all criteria for brain death have been met, cardiac asystole usually occurs within minutes after disconnecting the respirator; but if support is maintained, the heart may survive for much longer periods.
I. What constitute the minimum criteria to diagnose brain death
1. If the NE discloses no evidence of function after two NEs separated by 6 to 24 hours or, most conservatively, 72 hours, some physicians will then diagnose brain death. They consider the NE as the necessary and sufficient condition and do not require confirmatory laboratory tests.
2. Other physicians regard the NE as a necessary condition to diagnose brain death, but prefer one or both of two further safeguards (see, eg, the Pt of Ringel et al, 1988):
a. An isoelectric EEG, which shows no cerebral electrical activity.
b. Absence of cerebral blood flow as proven by radionuclide studies, conventional angiography, or transcranial Doppler ultrasound studies (Fig. 12-32).

FIGURE 12-32. Cerebral Death. The flow study (A) demonstrates prompt perfusion to the extracranial carotid system, but no intracranial arterial flow is seen at any time. The delayed images (B) demonstrate no visualization of the saggital sinus. The findings are consistent with cerebral death. (Courtesy of Robert H. Wagner, MD.)
3. These ancillary procedures prove useful if something prevents a full, adequate NE, such as severe burns of the face and head or traumatic destruction of facial parts. A radionuclide scan or EEG may be added to conclude the BDP after only one NE to facilitate organ transplantation rather than waiting for a prolonged period for a second clinical examination (Schneider and Ashwal, 1999).
4. The time of death is recorded as the time at which the Ex reaches a diagnosis of brain death.
J. Rationale for confirmatory tests and for involving more than one physician in the brain-death protocol
Involvement of at least three different physicians—the attending physician, the neurologist, and the radiologist or electroencephalographer—to diagnose brain death increases the probability that at least one of them will be honest, competent, and incorruptible. Then, if a pathologist does an autopsy, a fourth physician ensures the validity of the diagnosis. The pathologist by gross and microscopic examination can determine whether the brain had died many hours before removal, as distinguished from a brain subjected to prompt fixation shortly after death (Walker, 1985). The involved physicians do not meet as a committee and do not vote on whether the Pt’s brain is dead. They all ascertain independently of each other that brain-death criteria have been met, according to the techniques that each knows and thoroughly understands. With these safeguards, the diagnosis of brain death then becomes as infallible as humanly possible, and the process is protected from fraud, incompetence, or as a shield for murder.
K. Brain-death protocol in infants and children
Because of presumably greater resistance to hypoxia, the BDP for infants, in particular premature infants, is somewhat more conservative than for older Pts (Kohrman and Spivak, 1990; Schneider and Ashwal, 1999).
L. Pitfalls and precautions in the diagnosis of brain death by the neurologic examination
1. The two greatest pitfalls in the diagnosis of brain death are hypothermia and an overdose of depressant drugs. Rarely, a non-barbiturate drug may cause abolition of brainstem reflexes (Richard et al, 1998; Rizzo et al, 1993) as may toxic levels of aminoglycosides, anticholinergics, neuromuscular blocking agents, severe GBS (Hassan and Mumford, 1991). In severe pharmacologic depression or hypothermia, the NE may show no brain function, and the EEG may be isoelectric, yet the Pt may recover completely. Radionuclide scans or direct angiography will still demonstrate cerebral blood flow in these Pts who show no clinical or electrical evidence of brain function, but whose brains remain alive. And consider this scenario: a murderer who wanted to make a Pt appear brain dead could administer atropine to block pupillary and cardioinhibitory responses and curarize the Pt to block skeletomuscular responses. Such a Pt would have no effector responses and meet brain-death criteria on the purely clinical examination, but the EEG would demonstrate electrical activity and the radionuclide scan would demonstrate cerebral blood flow.
2. True decerebrate or decorticate posturing are inconsistent with the diagnosis of brain death. Some purely spinal reflexes remain in a large percentage of brain-dead Pts. The mechanism that caused brain death, such as cerebral edema or transforaminal herniation, may leave the spinal cord intact, whereas hypoxia-induced brain death may cause the cord to die also. With an intact cord, the Pt may show finger jerks, whole arm posturing, triple flexion, MSRs, and, extremely rarely, movements of the arms resembling decerebrate rigidity triggered by stimuli below the neck but not the face (Marti-Fabregas et al, 2000; Saposnik et al, 2000). Most dramatic of all is the “Lazarus sign” a complex movement of the upper limbs (spinal reflex) in which the arms flex and adduct, crossing the hands in front of the chest. The possibility of such ghastly movements that to the layman are totally mystifying and impossible to understand is one of the many reasons we always ask the family to remain in the waiting room during the examination.
3. By informing the family on a day-to-day basis of the Pt’s condition and prognosis, the physician encourages a rational response to the BDP. The family must understand that the purpose of the BDP is to recognize and verify beyond doubt that the person has already died and that continued support of the remaining organs is futile. Otherwise the family may misperceive termination of support as an effort to save money for an insurance company or the hospital, to provide organs for transplantation, or to commit euthanasia or genocide.
4. When all criteria for brain death have been rigidly fulfilled with respect to the NE and the appropriate ancillary studies (EEG and blood flow) have been correctly performed, the possibility for a mistake is virtually nil in Pts with mature brains. Because of different reactions of the immature and immature brains, the timing of the tests and some criteria for the BDP differ for infants (Schneider and Ashwal, 1999).
M. Permission for organ donation
Most physicians agree that no one on the organ transplant team should approach the family before completion of the BDP, because of the obvious conflict of interest. I think it best in general that the attending physician or neurologist raise this question after completion of the BDP and then introduce the representative from the transplant team.
N. Tabular summary of the neurologic examination of patients in coma, the persistent vegetative state, minimally conscious state, and locked-in syndrome (Table 12-8)
TABLE 12-8 • Neurologic Examination in Brain Death, Coma, PVS, Minimally Conscious State, and Locked-in Syndrome
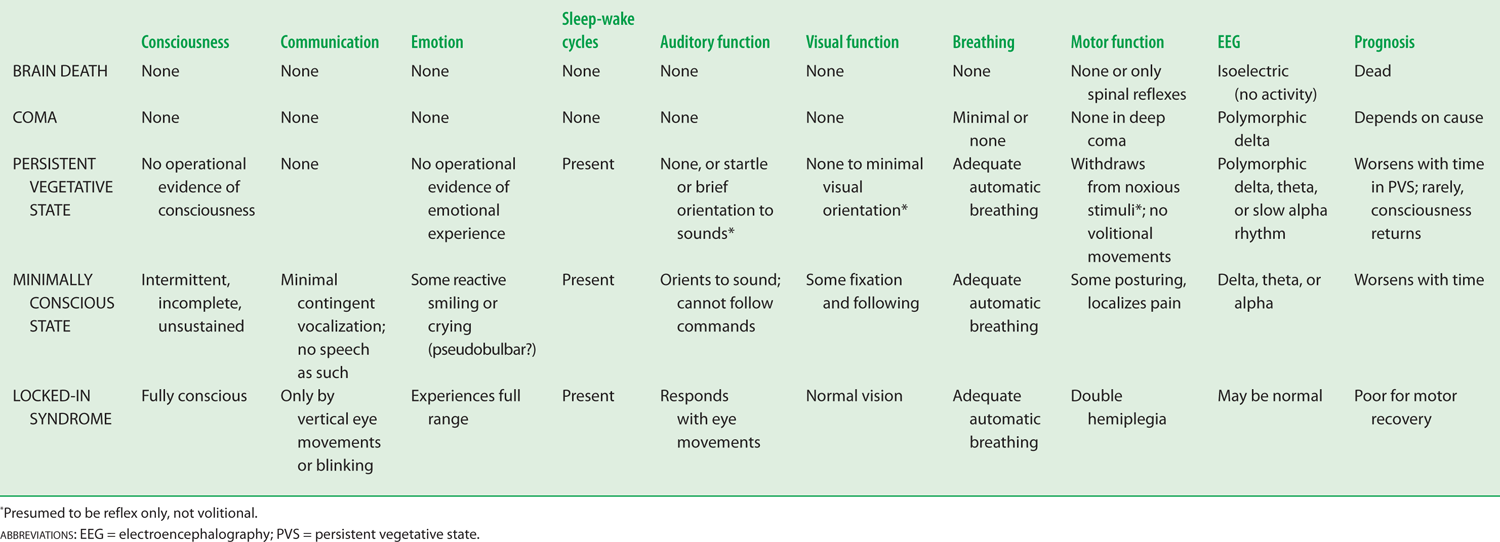
BIBLIOGRAPHY · The Neurologic Examination in the Diagnosis of Brain Death
American Academy of Neurology. Practice parameters for determining brain death in adults (summary statement). Neurology. 1995;45:1012–1014.
Facco E, Liviero MC, Munari M, et al. Short latency evoked potentials: new criteria for brain death. J Neurol Neurosurg Psychiatry. 1990;53(4):351–353.
Goudreau, Jl, Wijdicks EFM, Emery SF. Complications during apnea testing in the determination of brain death: predisposing factors. Neurology. 2000;55:1045–1048.
Gutmann DH, Marino PL. An alternative apnea test for the evaluation of brain death. Ann Neurol. 1991;30:852–853.
Hassan T, Mumford C. Guillain-Barré syndrome mistaken for brain death. Postgrad Med J. 1991;67:280–281.
Kohrman MH, Spivak BS. Brain death in infants: sensitivity and specificity of current criteria. Pediatr Neurol. 1990;6:47–50.
Marks SG, Zisfein J. Apneic oxygenation in apnea tests for brain death: a controlled trial. Arch Neurol. 1990;47:1066–1068.
Marti-Fabregas J, Lopez-Navidad A, Caballero F, et al. Decerebrate-like posturing with mechanical ventilation in brain death. Neurology. 2000;54:224–227.
Plum F, Posner J. Diagnosis of Stupor and Coma. 3rd ed. Philadelphia, PA: FA Davis; 1980.
Richard IH, Lapointe M, Wax P, et al. Non-barbiturate, drug induced reversible loss of brainstem reflexes. Neurology. 1998;51:639–640.
Ringel RA, Riggs, JE, Brick JF. Reversible coma with prolonged absence of pupillary and brainstem reflexes: an unusual response to a hypoxic-ischemia event in MS. Neurology. 1988;38:1275–1277.
Rizzo MA, Fisher M, Lock JP. Hypermagnesemic pseudo coma. Arch Int Med. 1993;153(9):1130–1132.
Saposnik G, Bueri JA, Maurino J, et al. Spontaneous reflex movements in brain death. Neurology. 2000;54:221–223.
Schneider S, Ashwal S. Determination of brain death in infants and children. In: Swaiman KE, Ashwal S, eds. Pediatric Neurology: Principles and Practice. 3rd ed. St. Louis, MO: Mosby; 1999, Chap. 62, 969–980.
Walker AD. Cerebral Death. 3rd ed. Baltimore, MD: Urban and Schwarzenberger; 1985.
Wijdicks EFM. The diagnosis of brain death. N Engl J Med. 2001a;344:1215–1221.
Wijdicks EFM, ed. Brain Death. Hagerston: Lippincott Williams & Wilkins; 2001b.
Wijdicks EFM. Brain death worldwide: accepted fact but no global consensus in diagnostic criteria. Neurology. 2002;58:20–25.
Zuckier L, Kolano J. Radionuclide studies in the determination of brain death: criteria, concepts, and controversies. Seminars in Nuclear Medicine. 2008;38(4);262–273.
X. THE NEUROLOGIC EXAMINATION IN INTERMITTENT DISTURBANCES OF CONSCIOUSNESS: SYNCOPE, SEIZURES, AND BLACKOUT SPELLS
A. Definition
Syncope means a sudden, brief lapse of consciousness, with loss of postural tone and spontaneous recovery, but not caused by epilepsy. Lay people call it fainting, falling out, or blacking-out. The medical investigation focuses on what precedes or triggers the syncope, either emotional factors or pathophysiologic events. Syncope is a common problem that annually affects 1 million Americans (Shukla and Zimetbaum, 2006) The incidence of organic syncope increases dramatically after the age of 70 years and almost doubles for Pts with cardiovascular disease. The sex incidence remains about equal (Soteriades et al, 2002). Figure 12-33 shows a classification of syncope.
FIGURE 12-33. Dendrogram for causes of syncope (reference only).
B. Pathophysiologic mechanisms of syncope
Whether triggered by emotional or organic causes, the final event that causes loss of consciousness is cerebral ischemia induced by several factors (Grubb and Olshansky, 1997; Lempert et al, 1994).
1. Vagal inhibition of the heart, causing bradycardia or asystole.
2. Loss of vasoconstrictor tone by sympathetic inhibition or insufficiency, causing hypotension. Sympathetic and parasympathetic discharges occur in syncope (Shen and Gersh, 1997).
3. Hydraulic or hypovolemic decreases in cerebral blood flow. The causes may be pooling of blood in the large veins, intrinsic heart disease with inadequate output, or occlusive vascular disease.
4. Cardiac dysrhythmias.
C. The medical history in syncope or any intermittent disorder such as headaches or seizures.
1. The three-phase description: For all intermittent clinical events, syncope, headaches, pain, and epilepsy divide the inquiry into preictal events, the ictus itself, and the postictal events or recovery period. Question family members who have seen an attack and view videos, if possible. Most important, learn the mode of onset. The clue to the cause comes from how the attack starts, not how it ends.
2. Preictal phase
a. What symptoms, posture, activities, and social circumstances prevail when the attacks come on? Is the Pt standing, sitting, moving, changing position, coughing, urinating, or hyperventilating?
b. Do the attacks all follow a pattern?
c. Does each attack start the same way? Ask the Pt: “What is the first change or warning of an attack?”
d. Is the Pt alone or with people, and, if with people, which ones?
e. Does the syncope evolve immediately or over minutes with premonitory feelings of fear, anxiety, impending doom, dizziness, and nausea?
3. The ictal phase
a. What happens during the attack: changes in breathing, falling, convulsive movements, sweating, pallor, cyanosis, and incontinence? Is the Pt injured? Is the tongue bitten?
b. Does the Pt lose consciousness? Ask: “Can you talk during the attack? Are you aware of what is going on?”
c. How long is the attack?
4. The postictal phase
a. How does the ictus end?
b. Is recovery sudden or gradual?
c. Does the Pt recognize that an ictus has occurred?
d. Is the Pt amnesic or disoriented?
e. Does the Pt regularly sleep afterward?
f. Is the Pt stiff and sore after an attack?
5. Reproducing the circumstances of the attack: Try to reproduce the circumstances that trigger an attack. If the Pt reports that a posture, such as head turning, elicits an attack, have the Pt turn the head. If hyperventilation or coughing precedes the attack, have the Pt hyperventilate or cough. If the Pt faints in the presence of his mother, observe him in the presence of his mother.
D. Observations during an attack
If the Ex has the fortune to witness an attack, have the Pt describe the symptoms—dizziness, weakness, nausea, flashing lights, etc. Protect the Pt during the unconscious phase.
1. Place the Pt flat on a soft surface.
2. Ensure that the Pt has an open airway. Turn the Pt’s head to the side to prevent aspiration in case of vomiting.
3. Check vital signs: blood pressure, pulse, respiratory rate, pupillary size and reaction, muscle tone, and reflexes.
4. Until you establish a diagnosis, draw a blood sample for hypoglycemia or hypocalcemia whenever you observe any attack that alters consciousness and consider giving intravenous glucose.
E. Psychiatric illness syncope (Fig. 12-33)
1. In psychiatric illness syncope, the Pt usually has a somatization disorder or is malingering. The Pt may experience weakness, sweating, nausea, abdominal discomfort, and sighing and show pallor at the onset, or it may be instantaneous.
2. Mental illness never manifests solely by syncope. It is only one shadow in the pattern of emotional turmoil (Leis et al, 1992).
3. Psychogenic attacks usually occur in the presence of people emotionally entangled with the Pt. At the end of the NE, call in the family to see how the family and Pt interact. The Pt may faint on the spot, thus clinching the diagnosis. Organic syncope is not “socially dependent.”
4. If Pts fall during the ictus, they usually do not hurt themselves or become incontinent. As in hysterical swaying on the Romberg test, the Pt may conveniently fall into the observer’s arms. In organic attacks, the fall often injures the Pt, but this distinction is by no means absolute.
5. Preceding an attack, some Pts hyperventilate. Always ask about changes in breathing and any associated symptoms (Hoefnagels et al, 1991) and do the hyperventilation test (pages 371-372). The test and an EEG are particularly important in the differential diagnosis of epilepsy (Stephenson, 2002).
F. Mechanical/hypovolemic syncope from inadequate return of blood to the heart (Fig. 12-33)
1. Orthostatic syncope (orthostatic hypotension)
a. Upon suddenly rising from a sitting or reclining position, the Pt feels giddy or may faint. The blood pressure has dropped rather than automatically adjusting to the erect position. Everyone experiences this phenomenon to some degree.
b. Take the blood pressure and pulse rate with the Pt reclining and upright. Normal young persons display an acceleration of the pulse by 5 to 25 beats and a drop in systolic blood pressure of no more than 25 mm Hg and an increase of up to 10 mm Hg in the diastolic pressure (Arnold et al, 1991). In association with symptoms, a consistent drop below 90 mm Hg in systolic pressure establishes orthostatic hypotension. The elderly show a lower blood pressure on standing than young persons. A tilt table test refines the analysis of orthostatic hypotension, and the Valsalva maneuver may provide further evidence of autonomic insufficiency (Almquist et al, 1989; Chen et al, 1989). Ask about medications that might cause hypotension.
2. Parade-ground syncope: If a person stands still for a period of time, as a soldier on a parade ground, blood pools in the inferior vena cava and in the lower extremities. After the person faints and becomes horizontal, blood returns to the circulation, allowing prompt recovery.
3. Micturition syncope: During urination, particularly with a very full bladder, the Pt faints (Lyle, 1961). The release of a large volume of urine from the bladder and the sudden relaxation of the abdominal wall drops abdominal pressure. Venous blood pools in the tributaries of the inferior vena cava, thus curtailing venous return to the heart. Also, cardioinhibitory reflexes may arise from the bladder. Similarly, the drop in abdominal pressure may cause syncope after rapid removal of ascitic fluid from the abdomen. Cardiac inhibition or vasodilation from loss of sympathetic vasoconstrictor tone may add to the mechanical effects in reducing cerebral blood flow.
4. Pregnancy syncope: When the pregnant woman lies on her back, the gravid uterus compresses the inferior vena.
5. Breath-holding spells
a. The most common syncope in children 6 to 48 months of age evolves through a stereotyped tetrad (DeMyer, 2002). The child may have literally dozens of these spells. Learn this tetrad.
i. Provocation by anger or frustration
ii. Expiratory apnea and cyanosis
iii. Unconsciousness and opisthotonic rigidity
iv. Stupor and somnolence
b. Notice that part (1) is the preictal stage, (2) and (3) are the ictus, and (4) is the postictal stage. The history is all important in establishing the triggering emotional event before each spell. The NE is normal. The mechanism may involve the compression of the vena cava by the prolonged forced expiration and vasovagal reflexes. The uncommon pallid type of breath-holding spell is more likely vasovagal (Shore and Painter, 2002).
6. Cough (tussive) syncope: The Pt, usually one with lung disease, has syncope after prolonged coughing. The syncope may result from a vagal reflex or from the Valsalva effect, both of which reduce the return of blood via the vena cava. In some cases, coughing or sneezing may precipitate syncope in Pts with the Chiari malformation (Corbett et al, 1976; Weig et al, 1991) or other posterior fossa lesions or with neck masses.
7. Stretch syncope: The Pt faints when extending the head while stretching. The syncope may result from a combination of the Valsalva maneuver, vasovagal mechanisms, and positional occlusion of the vertebral artery (Fig. 12-34; Pelekanos et al, 1990; Sturzenegger et al, 1995).
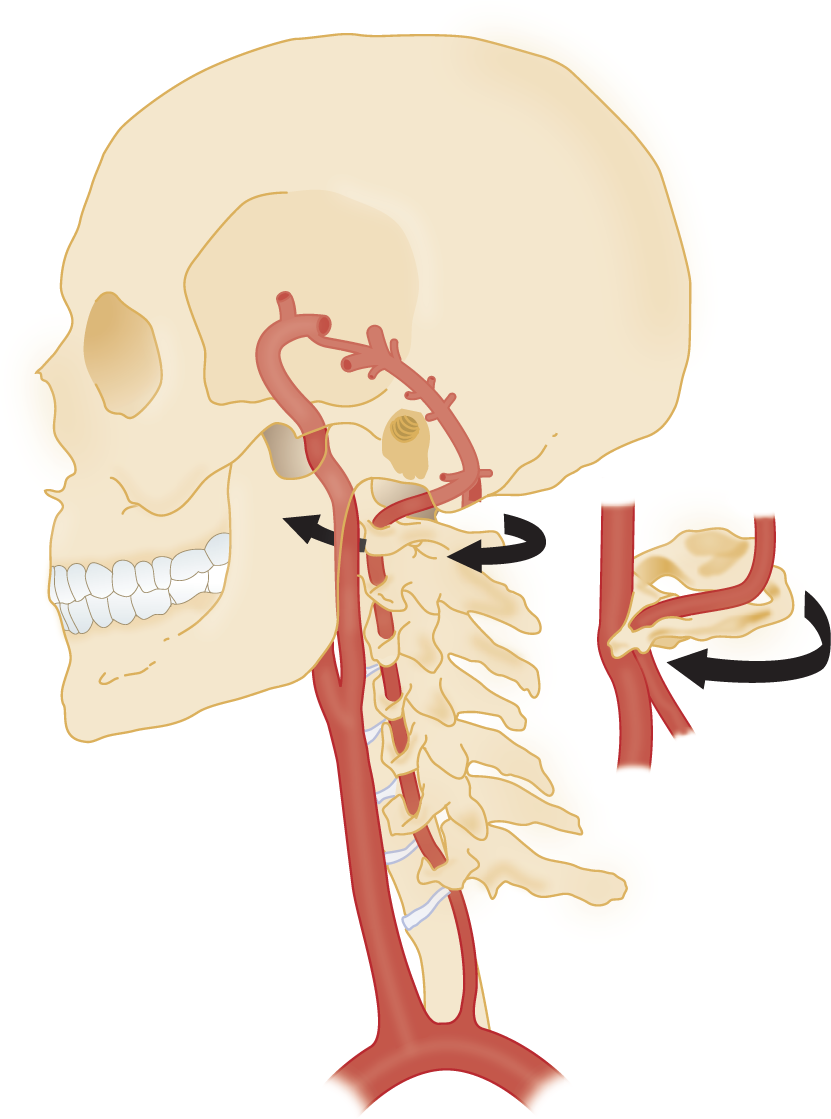
FIGURE 12-34. Diagram to show how turning of the head may kink or compress the carotid or vertebral arteries, thereby interfering with blood flow to the brain.
G. Neurogenic (vasovagal) syncope
1. Either a physical stimulus, such as coughing or head turning, or an emotional event, such as the sight of blood, triggers the syncope. In either case, the syncope has a pathophysiologic basis in bradycardia/hypotension acting through vasovagal mechanisms that inhibit the heart and vascular tone (Arnold et al, 1991). Cholinergic stimulation predominates, producing sweating, increased peristalsis, salivation, and bradycardia.
2. Syncope triggered by an emotional stimulus such as the sight of blood, a needle prick, or bad news may be called psychogenic vasovagal syncope. The interpretation and definition of this category, often called vasovagal syncope, differs from investigator to investigator (Landau and Nelson, 1996; Nahm and Freeman, 2001). Because these responses happen in otherwise normal persons, psychogenic vasovagal syncope, as defined here, differs from psychiatric illness syncope per se (Fig. 12-33).
H. Neurogenic vasovagal syncope, reflexogenic type, from swallowing, ear stimulation, or carotid sinus
1. Swallowing or glossopharyngeal syncope: The Pt faints when swallowing or in association with spontaneous pain in the ear and throat (glossopharyngeal neuralgia, analogous to trigeminal neuralgia).
2. Otogenic syncope: The Pt faints after stimulation of the external auditory canal, as during the insertion of an otoscope, or as a result of ear disease. Because the auditory canal and drum originate from the branchial arches, they receive sensory twigs from the branchial arch nerves, including CrNs IX and X. Thus, stimulation of the external auditory canal or drum may directly stimulate CrN IX or X afferents.
3. Reflexogenic syncope from carotid sinus hypersensitivity
a. Pathophysiology: Increased BP stimulates baroreceptors in the carotid sinus that compensate by causing vasodilation or cardiac inhibition. Hypersensitivity of the reflex causes syncope.
i. The carotid sinus is innervated by CrN _________
ii. Cardiac inhibition is a parasympathetic reflex with the efferent arc through CrN _________
iii. If carotid sinus hypersensitivity causes syncope by bradycardia/asystole, it is cardioinhibitory syncope (70%-75% of cases).
iv. If carotid sinus hypersensitivity causes syncope from inhibition of sympathetic vasoconstrictor tone, it is vasodepressor syncope (5%-10% of cases).
v. A third, controversial type of carotid sinus syncope is the cerebral type in which neither the pulse nor the blood pressure changes radically (Reese et al, 1962).
vi. A paradoxical reflex may inhibit the sympathetic outflow rather than causing the expected vasoconstriction and tachycardia. It may worsen hypotension from hemorrhagic shock and during isoproterenol infusion (Almquist et al, 1989).
b. Complete Table 12-9.
TABLE 12-9 • Mechanisms of Carotid Sinus Syncope
Type of carotid sinus syncope |
Mechanisms of syncope |
Vasodepressor |
|
Cardioinhibitory |
|
Cerebral |
(Decreased sympathetic vasoconstrictor tone)
(Reflex asystole or bradycardia) (Unknown)
c. Role of head turning and neck manipulation in syncope
i. A Pt with a hypersensitive carotid sinus or with swollen lymph nodes may faint when turning the head because of inadvertent mechanical stimulation of the carotid sinus.
ii. Head turning also may cause syncope by temporarily occluding a carotid or vertebral artery. In a normal young person, occlusion of a major cerebral artery may cause no symptoms or signs. In elderly, hypertensive, or arteriosclerotic Pts with occlusive arterial disease, the other arteries may fail to supply the brain adequately after occlusion of one (Fig. 12-34).
iii. Chiropractic neck manipulation may cause death or severe neurologic disability from vertebrobasilar infarction.
d. Indications for the carotid sinus massage test
i. A Pt with syncope precipitated by head turning or who has enlarged cervical lymph nodes.
ii. A Pt with lapses of consciousness for whom the history, physical examination, and other tests disclose no cause.
e. Contraindications to the carotid sinus massage test for carotid sinus sensitivity
i. Do not perform carotid sinus massage if patient has had an MI, stroke or transient ischemic attack in the previous three months.
ii. History of ventricular fibrillation, ventricular tachycardia, or carotid bruits, are relative contraindications.
f. Technique for the carotid sinus massage test
i. The technique for the carotid sinus massage has not been standardized. Some authors use carotid Doppler ultrasound to guide carotid sinus massage, and do not perform the maneuver if there is = 70% carotid artery stenosis.
ii. Complete the general physical and NE, including palpating the neck for nodes and listening over the head and neck for bruits. Monitor the Pt by electrocardiography (ECG) and preferably also EEG.
iii. Place the Pt sitting upright in a chair. A standing Pt may fall during the test or, if recumbent, may not have sufficient hypotension to faint, even though the Pt has a hypersensitive carotid sinus.
iv. Have the Pt repeat whatever head maneuver or position results in syncope.
v. The Ex must use a control test for suggestibility to exclude psychogenic syncope. State that you will rub the Pt’s neck, but do not suggest the outcome. Select any point on the neck away from the carotid sinuses and massage gently for 5 to 15 seconds. Fainting suggests psychogenic syncope.
vi. Locate the carotid sinus by gently palpating the carotid bifurcation. Press your fingers gently backward, just below the angle of the mandible and anterior to the sternocleidomastoid muscle. Record the blood pressure and pulse. Massage the carotid bulb with very gentle pressure for 5 to 15 seconds and again record the blood pressure and pulse. After 5 minutes, massage the other carotid bulb.
vii. In normal subjects, the pulse slows by about 15% (Arnold et al, 1991). Asystole lasting more than 3 s or a drop of more than 50 mm Hg in the systolic blood pressure indicates carotid sinus hypersensitivity. (Fujimura et al, 1989).
viii. Carotid sinus hypersensitivity can be treated by surgical denervation of the sinus or by parasympathetic blocking agents.
g. Name two mechanisms by which head turning can cause loss of consciousness.
_________
_________
h. What is the position of the Pt for the carotid sinus massage test? sitting/ standing/ reclining. Explain.
_________
_________sitting) (If standing, the Pt may fall. If reclining, the Pt may not faint, even with a hypersensitive carotid sinus.)
i. Similar considerations apply to the vasovagal reflex that follows ocular compression (Shore and Painter, 2002; Stephenson, 1990).
I. Vascular occlusive syncope
1. Carotid or vertebrobasilar transient ischemic attacks: Complete a thorough neurovascular examination. See Chapter 1, Section V B and Fig. 12-34 (Toole, 2010).
2. Subclavian steal syndrome: The Pt has stenosis, or occlusion, of the left subclavian artery proximal to the origin of the left vertebral artery. The Pt may have a bruit over the supraclavicular fossa and sometimes a thrill. The blood pressure in the left arm is reduced. Exercise of the left arm “steals” blood from the vertebral system, resulting in dizziness, blurred vision, long tract and cranial nerve, and other brainstem signs, including syncope (Fig. 12-35). Subclavian steal syndrome from Takayasu arteritis may rarely present with syncope.
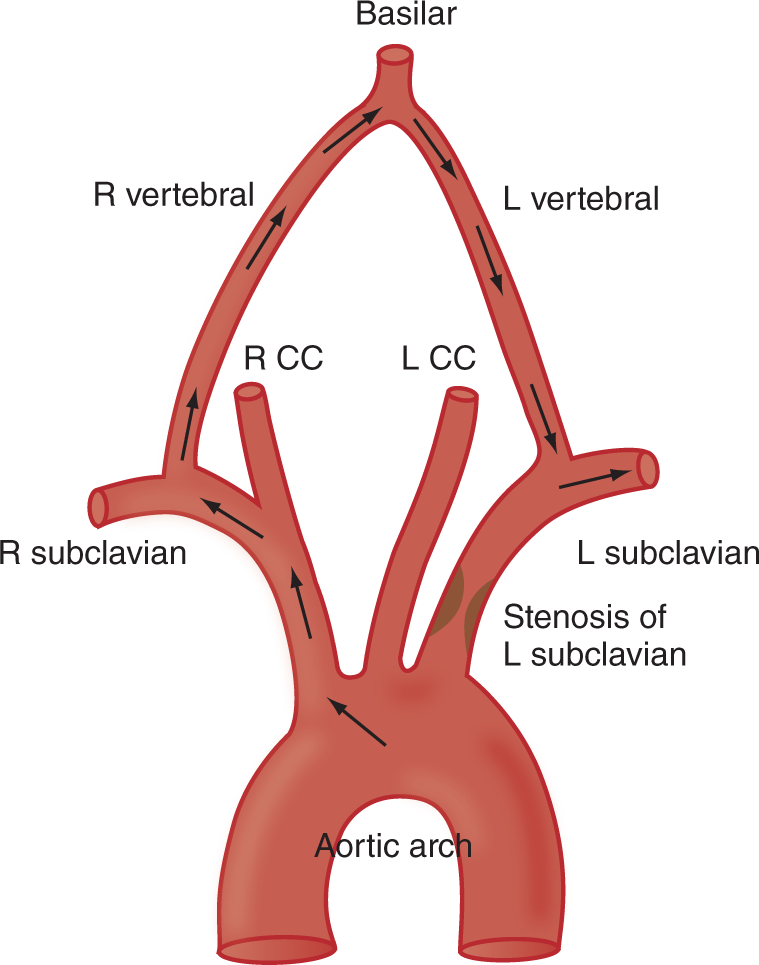
FIGURE 12-35. Diagram of the path of blood in the subclavian steal syndrome. Stenosis of the left subclavian artery reduces the pressure in the left subclavian artery, causing it to “steal” blood from the vertebral system (arrows). L = left; LCC = left common carotid artery; R = right; RCC = right common carotid artery.
J. Cardiac dysrhythmia syncope
This type of syncope results from a faulty impulse generation or transmission in the heart (Fujimura et al, 1989). Of special importance, look for Adams–Stokes atrioventricular block and for the prolonged QT interval syndrome, because its treatment prevents death. Syncope frequently requires an intensive cardiac and cardiovascular workup.
K. Epilepsy versus syncope
1. The most common differential diagnosis in syncope is epilepsy. Careful history and an epileptiform EEG generally distinguish epilepsy from syncope (Table 12-10).
TABLE 12-10 • Comparison of Neurologic Findings in Vasovagal/Hydraulic Hypotensive Syncope, Epilepsy, and Psychiatric Illness Syncopes
Vasovagal and hydraulic/hypotensive syncope |
Epilepsy |
Psychiatric illness syncope |
|
Age of onset |
Rare in infants |
Any age |
Adolescents/young adults (not in children) |
Sex |
About the same |
About the same |
Female > male |
Emotional instability |
Not correlated |
Not correlated |
Always present |
Trigger factors |
Standing after reclining, standing motionless, head postures, Valsalva maneuver |
Flashing lights, drug/alcohol withdrawal, hyperventilation, metabolic imbalance |
Presence of significant persons; emotionally trying events |
Preictal symptoms |
Weakness, dizziness, blurred vision, chest pain, sweating, abdominal discomfort |
Frequent aura, varying from hallucinations to abdominal discomfort |
Histrionic gestures and postures |
Length |
Seconds to minutes |
Seconds to minutes |
Brief to prolonged |
Autonomic signs |
Pallor, tachycardia, sweating |
Cyanosis, flushing |
None |
Blood pressure during attack |
Decreased |
Increased in motor seizures |
Normal |
Heart rate during episode |
Slow and forceful or rapid |
Rapid |
Normal |
Incontinence |
Rare |
Often |
Never |
Injury during attack |
Rare |
Frequent: bruises, tongue biting |
Almost never |
Postictal phase |
Prompt recovery with amnesia for duration of unconsciousness |
Confusion, retrograde/anterograde amnesia, sore muscles, hemiparesis, drowsiness |
May or may not have amnesia |
EEG during attack |
Diffuse slowing |
Epileptiform discharges |
Normal |
EEG between attacks |
Normal |
Abnormal usually |
Normal |
ABBREVIATION: EEG = electroencephalography. |
|||
2. Because cerebral ischemia disrupts brain metabolism, the Pt not only may faint but also may have a convulsion—an epileptic seizure—as part of an attack of syncope or breath-holding.
L. Subsequent workup of syncope when the initial evaluation fails to establish a diagnosis
The diagnosis of a single episode of syncope involves very different considerations from recurrent episodes of syncope. Syncope on exertion raises the question of cardiac output insufficiency. If the history and physical examination fail to establish the trigger factor for syncope, proceed with ECG and EEG or combined EEG, ECG, and video monitoring, or 24-hour ambulatory monitoring (Grubb and Olshansky, 1997; Low, 1997; Stephenson, 2002). Kapoor et al (2000) recommended ECG for most syncope Pts, but first be sure that you have exploited all the bedside tests. A tilt table test is indicated if the clinical findings suggest neurogenic vasovagal syncope. Occasionally, magnetic resonance imaging examination will disclose an unsuspected lesion, particularly of the posterior fossa, such as a Chiari type I malformation that causes the syncope (Weig et al, 1991). Despite a full workup, many Pts with blackout spells will remain undiagnosed after the initial evaluation (Kapoor et al, 1983; Soteriades et al, 2002). Above all, follow the Pt. The number of Pts with no diagnosis will drop as the follow-up visits continue.
BIBLIOGRAPHY · Syncope
Almquist A, Goldenberg IF, Milstein S, et al. Provocation of bradycardia and hypotension by isoproterenol and upright posture in patients with unexplained syncope. N Engl J Med. 1989;320:346–351.
Arnold RW, Dyer JA, Gould AB, et al. Sensitivity to vasovagal maneuvers in normal children and adults. Mayo Clin Proc. 1991;66:797–804.
Chen MY, Goldenberg AB, Milstein S, et al. Cardiac electrophysiologic and hemodynamic correlates of neurally mediated syncope. Am J Cardiol. 1989;63:66–72.
Corbett JJ, Butler AB, Kaufman B. “Sneeze syncope” basilar invagination and Arnold-Chiari type I malformation. J Neurol Neurosurg Psychiatry. 1976;39:381–384.
DeMyer W. Breath-holding spells. In: Maria BL, ed. Current Management in Child Neurology. London: BC Decker; 2002, Chap. 25, 321–323.
Fujimura O, Yee R, Klein GJ, et al. The diagnostic sensitivity of electrophysiologic testing in patients with syncope caused by transient bradycardia. N Engl J Med. 1989;321:1703–1706.
Grubb BP, Olshansky B. Syncope: mechanisms and management. Armon: Futura Publishing; 1997.
Hoefnagels WAJ, Padberg GW, Overweg J, et al. Syncope or seizure? The diagnostic value of the EEG and hyperventilation test in transient loss of consciousness. J Neurol Neurosurg Psychiatry. 1991;4:953–954.
Kapoor W, Karpf M, Wieland S, et al. A prospective evaluation and follow-up of patients with syncope. N Eng J Med. 1983;309:197–204.
Kapoor WN. Syncope. N Engl J Med. 2000;343:1856–1861.
Kaufman H, Saadia D, Voustianiouk A. Midodrine in neurally mediated syncope: a double-blind, randomized, crossover study. Ann Neurol. 2002;52:342–345.
Landau WM, Nelson DA. Clinical neuromythology XV. Feinting science: neurocardiogenic syncope and collateral vasovagal confusion. Neurology. 1996;46:609–618.
Leis AA, Ross MA, Summers AK. Psychogenic seizures: ictal characteristics and diagnostic pitfalls. Neurology. 1992;42:94–99.
Lempert T, Bauer M, Schmidt D. Syncope: a videometric analysis of 56 episodes of transient cerebral hypoxia. Ann Neurol. 1994;36:233–237.
Low P, ed. Clinical Autonomic Disorders. Philadelphia, PA: Lippincott-Raven; 1997.
Lyle C, Monroe JT, Flinn DE, et al. Micturition syncope: report of 24 cases. N Engl J Med. 1961;265:982–986.
Nahm F, Freeman R. Vasovagal syncope. The contributions of Sir William Gowers and Sir Thomas Lewis. Arch Neurol. 2001;58:509–511.
Pelekanos JT, Dooley JM, Camfield PR, et al. Stretch syncope in adolescence. Neurology. 1990;40:705–708.
Reese C, Green J, Elliott F. The cerebral form of carotid sinus hypersensitivity. Neurology. 1962;12:492–494.
Shen W, Gersh BJ. Fainting: appraoch to management. In Low PA, ed. Clinical Autonomic Disorders. 2nd ed. Philadelphia, PA: Lippincott-Raven; 1997, Chap. 48, 649–680.
Shore PM, Painter M. Adolescent asystolic syncope. J Child Neurol. 2002;17:395–397.
Shukla GJ, Zimetbaum PJ. Syncope. Cardiology patient page. Circulation. 2006;113:e715–e717.
Soteriades ES, Evans JC, Larson MG, et al. Incidence and prognosis of syncope. N Engl J Med. 2002;347:878–885.
Stephenson JBP. Fainting and syncope. In: Maria BL, ed. Current Management in Child Neurology. London: BC Decker; 2002, Chap. 56, 345–351.
Sturzenegger M, Newell DW, Douville CM, et al. Transcranial Doppler and angiographic findings in adolescent stretch syncope. J Neurol Neurosurg Psychiatry. 1995;58:367–370.
Toole’s Cerebrovascular Disorders. 6th ed. By E. Steve Roach, Kirsten Betterman, and Jose Biller, Cambridge University Press. 2010. Baltimore, MD: Lippincott Williams & Wilkins.
Weig SG, Buckthal PE, Choi SK, et al. Recurrent syncope as the presenting symptom of Arnold-Chiari malformation. Neurology. 1991;41:1673–1674.
 Learning Objectives for Chapter 12
Learning Objectives for Chapter 12