Lead
 Critical Values
Critical ValuesTest Explanation
Lead is a heavy metal found in the environment that is a heavy metal toxin. Although lead is now banned from household paints, it is still found in paint used before 1980. Lead is found in dirt from areas adjacent to homes painted with lead-based paints. Water transported through lead or lead-soldered pipe will contain some lead with higher concentrations found in water that is weakly acidic.
Lead inhibits aminolevulinic acid dehydratase and ferrochelatase, both of which catalyze synthesis of heme. The end result is decreased hemoglobin synthesis and anemia. Lead also is an electrophile that avidly forms covalent bonds with the sulfhydryl group of cysteine in proteins. Thus proteins in all tissues exposed to lead will have lead bound to them. The most common sites affected are epithelial cells of the gastrointestinal tract and epithelial cells of the proximal tubule of the kidney. The brain is also a common depository for excess lead.
Signs and symptoms in adults may include a decline in mental status, muscle weakness, headaches, memory loss, mood disorders, and miscarriage or premature birth in pregnant women. Children may demonstrate irritability, anorexia, weight loss, and learning difficulties.
Lead poisoning is a preventable condition that results from environmental exposure to lead. This exposure, indicated by elevated blood lead levels, can result in permanent damage of almost all parts of the body. However, its effects are most pronounced on the central nervous system and kidneys causing symptoms ranging from mild learning disabilities and behavioral problems to encephalopathy. Children less than 6 years of age are the most likely to be exposed and affected by lead. Blood lead levels are the best test for detecting and evaluating recent acute and chronic exposure. Blood lead samples are used to screen for exposure and to monitor the effectiveness of treatment. Lead in the human body can also be measured in blood, urine, bones, teeth, or hair. Blood tests are usually performed. Lead assay is performed on a quantitative inductively coupled plasma-mass spectrometer (hematofluorometry). If one test is elevated, it should be repeated.
At critically high levels, immediate medical evaluation is recommended and chelation therapy is considered when symptoms of lead toxicity are present. Although blood lead has the highest correlation with lead poisoning, lead can be detected in the urine, nails, and hair. These other specimens are used to corroborate blood analysis or document past lead exposure. If the hair is collected and segmented in a time sequence (based on length from root), the approximate time of exposure can be assessed.
Procedure and Patient Care
During
• Collect a venous blood in a royal blue-top tube. A tan-top (lead only) Becton-Dickinson tube can be used.
• A fingerstick can be performed to obtain nearly 1 mL of blood.
• In addition to the venous blood sample or the fingerstick, a few mL of EDTA whole blood can be collected.
• Usually the blood sample is sent to a central diagnostic laboratory. The results are available to the local hospital in 7 to 10 days.
Legionnaires Disease Antibody
Indications
This test is indicated in patients suspected to have Legionnaires disease and who have negative cultures and smears identifying Legionella.
Test Explanation
Legionnaires disease was originally described as a fulminating pneumonia caused by Legionella pneumophila, a tiny, gram-negative, rod-shaped bacterium. Nearly half of the clinical cases have been caused by serogroup type 1. This organism can also cause an influenza type of illness called “Pontiac fever.”
The diagnosis of Legionnaires disease can be made by culturing this organism from suspected infected fluid, such as blood, sputum, or pleural fluid, or from lung tissue. Sputum for this test is best obtained by transtracheal aspiration or from bronchial washings. However, growing this organism in culture is difficult. A negative culture does not mean that the patient does not have Legionnaires disease. Another method of diagnosis is by directly identifying the organism in a microscopic smear of infected fluid with the use of direct fluorescent antibody methods. If positive, this allows for rapid identification of Legionella. However, this is difficult also because the concentration may not be high enough to see the bacterium in the specimen.
The most common and easiest method for diagnosis is detection of the antibody directed against the Legionnaires bacterium in the patient's blood. This is done if the culture or direct fluorescent tests are negative. The indirect fluorescent antibody assay or enzyme-linked immunosorbent assay (ELISA) methods are commonly used. A presumptive diagnosis of Legionnaires disease can be made in a symptomatic person when a single antibody titer is 1:256 or greater. Another way to make the diagnosis is to perform the antibody test 1 and 3 weeks after the onset of symptoms. A fourfold rise in titer to at least 1:128 between the acute (1-week) and the convalescent (3-week) phases is diagnostic. Unfortunately it may take 4 to 6 weeks for serologic tests to be positive. The patient would be seriously ill by then. Legionella antigens in the urine may be identified a few days after the onset of the clinical symptoms, but the sensitivity is very low (about 30%).
Leucine Aminopeptidase (LAP, Aminopeptidase Cytosol)
Indications
This test is used for diagnosing liver disorders. It aids in the differential diagnosis of patients with high levels of alkaline phosphatase.
Test Explanation
LAP is an intracellular enzyme that exists in the hepatobiliary system and, to a much smaller degree, in the pancreas and the small intestine. When disease or injury affects those organs, the cells lyse and LAP is spilled out into the bloodstream. Produced almost exclusively by the liver, LAP is used in diagnosing liver disorders and in the differential diagnosis of increased levels of alkaline phosphatase (ALP). LAP levels tend to parallel ALP levels in hepatic disease. LAP is a sensitive indicator of cholestasis; however, unlike ALP, LAP remains normal in bone disease. LAP can be detected in both the blood and the urine. Patients with elevated serum LAP levels will show elevations in urine levels. When the urine LAP level is elevated, however, the blood level may have already returned to normal.
Interfering Factors
• Pregnancy may cause increased values if tested by the enzyme method. Although there is not a quantitative increase in this “LAP-like” enzyme, its activity is increased. This causes a false increase in the LAP if tested by the enzyme method.
 Drugs that may cause increased LAP levels include estrogens and progesterones.
Drugs that may cause increased LAP levels include estrogens and progesterones.
Test Results and Clinical Significance
 Increased Levels
Increased Levels
Hepatobiliary disease (e.g., hepatitis, cirrhosis, hepatic necrosis, hepatic ischemia, hepatic tumor, hepatotoxic drugs, cholestasis, gallstones): LAP is an enzyme that exists in the liver and biliary cells. Disease or injury of these tissues will cause the cells to lyse. LAP will spill out into the bloodstream, and levels will rise.
Related Tests
Creatine Phosphokinase (CPK) (p. 186). This enzyme is used similarly to aspartate aminotransferase (AST) and exists predominantly in heart and skeletal muscle.
Alanine Aminotransferase (ALT) (p. 39). This enzyme is used similarly to AST and exists predominantly in the liver.
Lactic Dehydrogenase (LDH) (p. 329). This is an intracellular enzyme used to support the diagnosis of injury or disease involving the heart, liver, red blood cells (RBCs), kidneys, skeletal muscle, brain, and lungs.
Aspartate Aminotransferase (AST) (p. 119). This is another enzyme existing predominantly in the liver.
Gamma-Glutamyl Transpeptidase (GGTP) (p. 246). This is another enzyme that exists predominantly in the liver.
Alkaline Phosphatase (p. 47). This is another enzyme existing predominantly in the liver.
5'-Nucleotidase (p. 376). This is another enzyme existing predominantly in the liver.
Lipase
Test Explanation
The most common cause of an elevated serum lipase level is acute pancreatitis. Lipase is an enzyme secreted by the pancreas into the duodenum to break down triglycerides into fatty acids. As with amylase, lipase appears in the bloodstream following damage to or disease affecting the pancreatic acinar cells.
Because lipase was thought to be produced only in the pancreas, elevated serum levels were considered to be specific to pathologic pancreatic conditions. It is now apparent that other conditions can be associated with elevated lipase levels. Lipase is excreted through the kidneys. Therefore elevated lipase levels are often found in patients with renal failure. Intestinal infarction or obstruction also can be associated with lipase elevation. However, the lipase elevations in nonpancreatic diseases are less than three times the upper limit of normal as compared with pancreatitis, in which they are often 5 to 10 times normal values. Other conditions such as cholangitis, mumps, cholecystitis, or peptic ulcer are more rarely associated with elevated lipase levels.
In acute pancreatitis, elevated lipase levels usually parallel serum amylase levels. The lipase levels usually rise a little later than amylase levels (24 to 48 hours after the onset of pancreatitis) and remain elevated for 5 to 7 days. Because they peak later and remain elevated longer than the serum amylase levels, serum lipase levels are more useful in the late diagnosis of acute pancreatitis. Lipase levels are less useful in more chronic pancreatic diseases (e.g., chronic pancreatitis, pancreatic carcinoma).
Interfering Factors
Drugs that may cause increased lipase levels include bethanechol, cholinergics, codeine, indomethacin, meperidine, methacholine, and morphine.
 Clinical Priorities
Clinical Priorities
• This test is useful in evaluating pancreatitis. Lipase elevations are often 5 to 10 times normal values in pancreatitis.
• In acute pancreatitis, elevated lipase levels usually parallel serum amylase levels. Because lipase levels peak later and remain elevated longer than amylase levels, they are more useful in the late diagnosis of acute pancreatitis.
Test Results and Clinical Significance
Increased Levels
Pancreatic diseases (e.g., acute pancreatitis, chronic relapsing pancreatitis, pancreatic cancer, pancreatic pseudocyst): Lipase exists in the pancreatic cell and is released into the bloodstream when disease or injury affects the pancreas.
Biliary diseases (e.g., acute cholecystitis, cholangitis, extrahepatic duct obstruction): Although the pathophysiology of these observations is not well understood, it is suspected that lipase exists inside the cells of the hepatobiliary system. Disease or injury of these tissues would cause the lipase to leak into the bloodstream and cause levels to be elevated.
Renal failure: Lipase is excreted by the kidney. If excretion is poor, as in renal failure, lipase levels will rise.
Intestinal diseases (e.g., bowel obstruction, infarction): Lipase exists in the mucosal cells lining the bowel (mostly in the duodenum). Injury through obstruction or ischemia will cause the cells to lyse. Lipase will leak into the bloodstream and cause levels to be elevated.
Salivary gland inflammation or tumor: Like amylase, salivary glands contain lipase, although to a much lesser degree. Tumors, inflammation, or obstruction of salivary ducts will cause the cells to lyse. Lipase will leak into the bloodstream and cause levels to be elevated.
Peptic ulcer disease: The pathophysiology of this observation is not well understood. Certainly, in perforated peptic disease the lipase in the gastrointestinal (GI) contents leaks out into the peritoneum, where it is picked up by the bloodstream. Lipase levels rise.
Related Test
Amylase (p. 61). Disease affecting the pancreas also will cause elevations of this enzyme.
Lipoprotein-Associated Phospholipase A2 (Lp-PLA2, PLAC Test)
Test Explanation
Lipoprotein-associated phospholipase A2 (Lp-PLA2) promotes vascular inflammation through the hydrolysis of oxidized LDL within the intima, contributing directly to the atherogenic process. Lp-PLA2 is an independent predictor of cardiovascular disease. When combined with CRP (p. 184), testing for Lp-PLA2 markedly increases the predictive value in determining risk for a cardiac event, especially in patients whose Adult Treatment Panel III (ATP III) cardiac risks are moderate. Lp-PLA2 levels >200 ng/mL would warrant reclassifying the patient to the next highest ATP risk category, which would require more aggressive use of cholesterol-lowering agents. Lp-PLA2 may play an important role in the progression of atherosclerosis and overall plaque stability. Lp-PLA2 may be an effective target for anti-atheromatous therapies in the future.
Lp-PLA2 is also an accurate aid in assessing the risk for ischemic stroke associated with atherosclerosis at all levels of blood pressure. The PLAC test is an enzyme-linked immunosorbent assay (ELISA) using two highly specific monoclonal antibodies to measure the level of Lp-PLA2 in the blood.
Lipoproteins (High-Density Lipoproteins [HDL, HDL-C] Low-Density Lipoproteins [LDL, LDL-C], Very Low–Density Lipoproteins [VLDL], Lipoprotein Electrophoresis, Lipoprotein Phenotyping, Lipid Fractionation, Non-HDL Cholesterol, Lipid Profile)

Indications
Lipoproteins are considered to be an accurate predictor of heart disease. As part of the lipid profile, these tests are performed to identify persons at risk for developing heart disease and to monitor therapy if abnormalities are found (Box 2-14).
Test Explanation
Lipoproteins are proteins in the blood whose main purpose is to transport cholesterol, triglycerides, and other insoluble fats. They are used as markers indicating the levels of lipids within the bloodstream. Lipoproteins can be classified by their measured density.
General categories of lipoproteins, listed in order from larger and less dense (more fat than protein) to smaller and denser (more protein, less fat) are as follows:
• Chylomicrons—carry triacylglycerol (fat) from the intestines to the liver, skeletal muscle, and to adipose tissue.
• Very low–density lipoproteins (VLDL)—carry (newly synthesized) triacylglycerol from the liver to adipose tissue.
• Intermediate-density lipoproteins (IDL)—are intermediate between VLDL and LDL. They are not usually detectable in the blood.
• Low-density lipoproteins (LDL)—carry cholesterol from the liver to cells of the body. Sometimes referred to as the “bad cholesterol” lipoprotein.
• High density lipoproteins (HDL)—collects cholesterol from the body's tissues (and vascular endothelium) and brings it back to the liver. Removing lipids from the endothelium (reverse cholesterol transport) provides a protective effect against heart disease. Therefore HDL is referred to as the “good cholesterol” lipoprotein.
The “lipid profile” usually measures total cholesterol (discussed separately on p. 154), triglycerides, HDL, LDL, and VLDL. Through the use of segmented gradient gel electrophoresis (SGGE), lipoproteins could be subclassified to more accurately indicate cardiovascular risks and familial risks of heart disease. Levels of lipoproteins are genetically influenced; however, these levels can be altered by diet, lifestyle, and medications.
Clinical and epidemiologic studies have shown that total HDL cholesterol is an independent, inverse risk factor for coronary artery disease (CAD). Low levels (<35 mg/dL) are believed to increase a person's risk for CAD, while high levels (>60 mg/dL) are considered protective. When HDL and total cholesterol measurements are combined in a ratio fashion (Table 2-36), the accuracy of predicting CAD is increased. The total cholesterol/HDL ratio should be at least 5:1, with 3:1 being ideal.
TABLE 2-36
Risk for Coronary Heart Disease Based on Ratio of Cholesterol to High-Density Lipoproteins
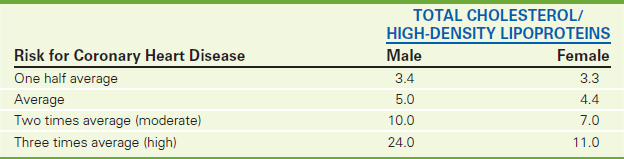
SGGE identified five subclasses of HDL (2a, 2b, 3a, 3b, and 3c), but only 2b is cardioprotective. HDL 2b is the most efficient form of HDL in reverse cholesterol transport. Patients with low total HDL levels often have low levels of HDL 2b. When levels of total HDL are between 40 and 60, cardioprotective levels of HDL 2b are minimal. However, when levels of total HDL are greater than 60, levels of HDL 2b predominate, and efficient reverse cholesterol transport takes place. This protects the coronary arteries from disease. The other subclasses of HDL are not capable of reverse cholesterol transport and therefore are not cardioprotective. Levels of HDL 2b can be increased by niacin supplements but not by statins (i.e., HMG-CoA reductase inhibitors [simvastatin, lovastatin]).
LDLs (“bad” cholesterol) are also cholesterol rich. However, most cholesterol carried by LDLs can be deposited into the lining of the blood vessels and is associated with an increased risk for arteriosclerotic heart and peripheral vascular disease. Therefore high levels of LDLs are atherogenic. Target LDL levels vary according to the risk profile of the patient (Table 2-37). For example, the optimal LDL level should be less than 70 mg/dL in patients at high risk for heart disease. The LDL level can be calculated using a modified Friedwald formula:
TABLE 2-37
National Cholesterol Education Program Therapy 2004 Guidelines for Low-Density Lipoproteins
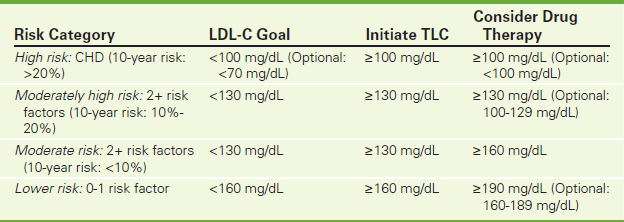
CHD, Coronary heart disease; LDL-C, low density lipoprotein cholesterol; TLC, therapeutic lifestyle changes (reduced intake of saturated fats and cholesterol, drug therapy, increased fiber, weight reduction, and increased physical activity).
Risk factors: cigarette smoking, hypertension, low HDL cholesterol, family history, and age.
10-year risk: Data from the Framingham Heart Study used to estimate risk of CHD (age, gender, HDL cholesterol, total cholesterol, systolic B/P, use of B/P medications).

There are other formulas for deriving LDL, which may account for different sets of normal values. The formula is inaccurate if the triglycerides exceed 400 mg/dL. More recently, laboratory chromogenic methods in which various detergents are used to separate out LDL allow for a more accurate measurement of LDL. This method uses a unique detergent to solubilize only the non-LDL lipoprotein particles and a second detergent solubilizes the remaining LDL particles, which are then measured by a chromogenic coupler that provides color formation.
With the use of SGGE, LDL has been divided into seven classes based on particle size. These subclasses include (from largest to smallest) LDL I, LDL IIa, LDL IIb, LDL IIIa, LDL IIIb, LDL IVa, and LDL IVb. The most commonly elevated forms of LDL (IIIa and IIIb) are small enough to get between the endothelial cells and cause atheromatous disease. The larger LDL particles (LDL I, LDL IIa, and LDL IIb) cannot get into the endothelial layer and therefore are not associated with increased risk for disease. LDL IVa and IVb, however, are very small and are associated with aggressive arterial plaques that are particularly vulnerable to ulceration and vascular occlusion. Nearly all patients with levels of LDL IVa and IVb greater than 10% of total LDL have vascular events within months.
LDL patterns can be identified, and they are associated with variable risks of coronary artery disease (CAD). LDL pattern A is seen in patients with mostly large LDL particles and does not carry increased risks for CAD. LDL pattern B is seen in patients with mostly small LDL particles and is associated with an increased risk for CAD. An intermediate pattern is noted in a large number of patients; they have small and large LDL particles and experience an intermediate risk for CAD. LDL levels can be lowered with diet, exercise, and statins.
Because LDL particles vary in size and composition, the amount of cholesterol they carry (LDL-C) is not a reliable measure of the number of LDL particles (LDL-P) and a patient's risk for CHD. Direct measurement of the number of LDL particles (i.e., LDL-P) by Nuclear Magnetic Resonance (NMR) Spectroscopy provides prognostic information that is independent of LDL-C. Direct measurement by LDL-P has proved to be a better predictor of CHD events than LDL-C.
VLDLs, though carrying a small amount of cholesterol, are the predominant carriers of blood triglycerides. To a lesser degree, VLDLs are also associated with an increased risk for CAD because they can be converted to LDL by lipoprotein lipase in skeletal muscle. The VLDL value is sometimes expressed as a percentage of total cholesterol. Levels in excess of 25% to 50% are associated with increased risk for coronary disease.
The Adult Treatment Panel III (ATP III) of the National Cholesterol Education Program issued an evidence-based set of guidelines on cholesterol management. The goal for high-risk patients (those with known coronary artery disease or > 2 risk factors) is an LDL lower than 70 mg/dL. All ATP reports have identified low-density lipoprotein cholesterol (LDL-C) as the primary target of cholesterol lowering therapy. Many prospective studies have shown that high serum concentrations of LDL-C are a major risk factor for coronary heart disease (CHD). Moreover, lowering LDL-C levels will reduce the risk for major coronary events.
The World Health Organization adopted the Fredrickson classification of lipid disorders to identify particular lipoprotein patterns (phenotypes) that are associated with certain inherited or acquired diseases or syndromes. Fredrickson's classification (Table 2-38), through the use of electrophoresis, simply identifies which lipoproteins are raised.
TABLE 2-38
Primary Hyperlipidemias (WHO/Fredrickson Classification)
| Fredrickson Classification | Elevated Lipoprotein | Associated Clinical Disorders |
| I | Chylomicrons | Lipoprotein lipase deficiency, apolipoprotein CII deficiency, uncontrolled diabetes mellitus (DM) |
| IIa | LDL | Familial hypercholesterolemia, nephrosis, hypothyroidism, familial combined hyperlipidemia |
| IIb | LDL, VLDL | Familial combined hyperlipidemia |
| III | Intermediate-density lipoproteins | Dysbetalipoproteinemia, DM, alcoholism |
| IV | VLDL | Familial hypertriglyceridemia, familial combined hyperlipidemia, diabetes mellitus |
| V | Chylomicrons, VLDL | Diabetes, nephrosis, malnutrition |
There are a variety of methods used to measure the lipoprotein classes. All require serum separation, usually by ultracentrifugation. In the past, lipoproteins were measured through the use of electrophoresis. Immunologic, catalase reagent, and chemical kits are now available for accurately quantifying lipoproteins.
Interfering Factors
• Smoking and alcohol ingestion decrease HDL levels.
• Binge eating can alter lipoprotein values.
• HDL values are age- and sex-dependent.
• HDL values, like cholesterol, tend to decrease significantly for as long as 3 months following myocardial infarction (MI).
• HDL is elevated in patients with hypothyroid and diminished in those with hyperthyroid.
• High triglyceride levels can make LDL calculations inaccurate.
Clinical Priorities
• Lipoproteins are considered to be predictors of heart disease. Blood levels should be collected after a 12- to 14-hour fast.
• HDL is often called good cholesterol, because it removes cholesterol from the tissues and transports it to the liver for excretion. High levels are associated with a decreased risk for coronary heart disease.
• LDL is often called bad cholesterol, because it carries cholesterol and deposits it into the peripheral tissues. High levels are associated with an increased risk for CHD.
Test Results and Clinical Significance
 Decreased HDL
Decreased HDL
Metabolic syndrome: This syndrome is associated with an atherogenic lipid profile (ALP) that includes decreased HDL, increased triglycerides, elevated fasting glucose, high blood pressure, and abdominal obesity measured by waist circumference.
Familial low HDL: Genetically, the patient is predetermined to have low HDL levels. As a result, these patients are at high risk for CHD.
Hepatocellular disease (e.g., hepatitis, cirrhosis): HDL is made in the liver. Without liver function, HDL is not made and levels fall.
Hypoproteinemia (e.g., nephrotic syndrome, malnutrition): With loss of proteins, HDL is not made and levels fall. When the hypoproteinemia is severe, however, and oncotic pressures fall, the production of lipoproteins could be stimulated and actually rise. Elevation of HDL occurs only late in the disease.
Increased LDL and VLDL
Familial LDL lipoproteinemia: Genetically, the patient is predetermined to have high LDL levels.
Nephrotic syndrome: The loss of proteins diminishes the plasma oncotic pressures. This appears to stimulate hepatic lipoprotein synthesis of LDL and possibly to diminish lipoprotein disposal of the same.
Glycogen storage diseases (e.g., von Gierke disease): VLDL synthesis is increased and excretion is diminished. VLDL and LDL levels rise.
Hypothyroidism: VLDL and LDL catabolism is diminished. VLDL and LDL levels rise. This is a common cause of lipid abnormalities, especially among women.
Alcohol consumption: Hyperlipidemias are known to occur in persons who drink excessive quantities of alcohol. However, there also may be a genetic factor associated with this observation.
Chronic liver disease (e.g., hepatitis, cirrhosis): The liver catabolizes LDL. Without that catabolism, blood levels increase.
Hepatoma: The normal inhibition of LDL synthesis by eating dietary fats does not occur. LDL synthesis continues unabated. LDL levels rise.
Gammopathies (e.g., multiple myeloma): High levels of gamma globulins (IgG and IgM) attach to the VLDL and LDL molecule and thereby decrease their catabolism.
Familial hypercholesterolemia type IIa: LDL receptors are altered, and LDL is produced at increased rates.
Cushing syndrome: VLDL synthesis is increased. VLDL is converted to LDL.
Apoprotein CII deficiency: This genetic defect is associated with a deficiency of lipoprotein lipase. As a result, VLDL and other lipoproteins (chylomicrons) accumulate.
Decreased LDL and VLDL
Familial hypolipoproteinemia: Genetically, the patient is predetermined to have low VLDL or LDL levels.
Hypoproteinemia (e.g., malabsorption, severe burns, malnutrition): Early in the course of this process, LDLs are low. However, later the LDL and VLDL levels can actually rise.
Hyperthyroidism: Catabolism of LDL and VLDL is increased and levels fall.
Related Tests
Cholesterol (p. 154). This is a measure of total cholesterol in the blood. It is a part of the lipid profile.
Triglycerides (p. 504). This is a measure of total triglyceride in the blood. It is a part of the lipid profile.
Apolipoproteins (p. 106). This test is used to measure apolipoprotein levels. This may be a better indicator of atherogenic risks than total HDL or total LDL.
Luteinizing Hormone (LH, Lutropin) and Follicle-Stimulating Hormone (FSH) Assay

Indications
LH/FSH levels are helpful in the determination of menopause. Furthermore, they are integral in the evaluation of suspected gonadal failure. Infertility evaluations also include these tests.
Test Explanation
LH and FSH are glycoproteins produced in the anterior pituitary gland in response to stimulation by gonadotropin-releasing hormone (GNRH), previously called luteinizing-releasing hormone. GNRH is stimulated when circulating levels of estrogen (in females) or testosterone (in males) are low. Through a feedback mechanism, GNRH is stimulated by the hypothalamus, which in turn stimulates the production and release of LH and FSH. These two hormones then act on the ovary or testes. In the female, FSH stimulates the development of follicles in the ovary. In the male, FSH stimulates Sertoli cell development. In the female, LH stimulates follicular production of estrogen, ovulation, and formation of a corpus luteum. In the male, LH stimulates testosterone production from the Leydig cells. In the end, estrogen or testosterone is produced, which in turn inhibits FSH and LH. FSH is necessary for maturation of the ovaries and testes. FSH and LH are necessary for sperm production. In the female these hormones are secreted differently at different times in the menstrual cycle. The midcycle peak of FSH is necessary for follicle/ovum formation. LH also must peak about that same time to stimulate ovulation or corpus luteal formation that could potentially support an embryo if fertilization were to occur.
Earlier bioassays could not distinguish FSH from LH. Therefore they were often measured together. For that matter, early bioassays often included thyroid-stimulating hormone and human chorionic gonadotropin. Now with the use of better methodology such as quantitative electrochemiluminescent or liquid chromatography–tandem mass spectrometry, these hormones can each be measured separately and accurately.
LH is secreted in a pulsatile manner. One specimen may not accurately indicate total body levels of this hormone. Often several specimens of blood are obtained 20 to 30 minutes apart, and the blood is pooled or results of each are averaged. The variable nature of LH can be diminished by measuring LH in a 24-hour urine sample. The disadvantage is that LH values can be falsely low because of dilution with large urine volumes. Spot urine tests have become very useful in the evaluation and treatment of infertility. Because LH is rapidly excreted into the urine, the plasma LH surge that precedes ovulation by 24 hours can be recognized quickly and easily. This is used to indicate the period when the woman is most fertile. The best time to obtain a urine specimen is between 11 AM and 3 PM. Usually the woman begins to test her urine on the 10th day following the onset of her menses and continues to do so daily. Home kits using a color change as an end point are now marketed to make this process even more convenient.
These hormones are used in the evaluation of infertility. Performing an LH assay is an easy way to determine if ovulation has occurred. An LH surge in blood levels indicates that ovulation has taken place. Under the influence of LH, the corpus luteum develops from the ruptured Graafian follicle. Daily samples of serum LH around the middle of the woman's cycle can detect the LH surge, which is thought to occur on the day of maximal fertility.
These assays (particularly FSH) also determine whether a gonadal insufficiency is primary (problem with the ovary/testicle) or secondary (caused by pituitary insufficiency resulting in reduced levels of FSH and LH). Elevated levels of FSH and LH in patients with gonadal insufficiency indicate primary gonadal failure, as may be seen in women with polycystic ovaries or during menopause. In secondary gonadal failure, LH and FSH levels are low as a result of pituitary failure or some other pituitary-hypothalamic impairment, stress, malnutrition, or physiologic delay in growth and sexual development.
FSH and LH assays are often done to diagnose menopause. LH hormones are also used to study testicular dysfunction in men and to evaluate endocrine problems related to precocious puberty in children. The use of these hormone assays can also help in the evaluation of disorders of sexual differentiation, such as Klinefelter syndrome.
Interfering Factors
• Recent use of radioisotopes may affect test results if the testing method is performed by radioimmunoassay. The previously administered radioisotope may interfere with the results.
• Human chorionic gonadotropin (hCG) and thyroid-stimulating hormone (TSH) may interfere with some immunoassay methods because of the similarities of part of the hormone molecule. Therefore patients with hCG-producing tumors and those with hypothyroid should be expected to have falsely high LH levels.
Drugs that may increase LH or FSH levels include anticonvulsants, cimetidine, clomiphene, digitalis, levodopa, naloxone, and spironolactone.
Drugs that may decrease LH levels include digoxin, estrogens, oral contraceptives, progesterones, steroids, testosterone, and phenothiazines.
Clinical Priorities
• Levels of FSH and LH vary in the female patient according to phases in the menstrual cycle.
• These hormones are valuable in the evaluation of infertility. Daily samples of LH around a woman's midcycle can detect the LH surge, which is thought to occur on the day of maximum fertility.
• Spot urine tests have become useful in evaluating and treating infertility. Home test kits are now available for detecting LH in the urine.
• FSH and LH assays are often performed to diagnose menopause so hormone replacement can be started.
Procedure and Patient Care
During
Urine
• Collect a 24-hour specimen; a preservative may be used.
• Keep the specimen refrigerated during the collection period.
• Note that the patient may also perform LH assays at home using a home urine test or a 24-hour urine test.
• If a spot urine test is performed, follow the directions accompanying the kit.
Test Results and Clinical Significance
Increased Levels
Gonadal failure (e.g., physiologic menopause, ovarian dysgenesis [Turner syndrome], testicular dysgenesis [Klinefelter syndrome], castration, anorchia, hypogonadism, polycystic ovaries, complete testicular feminization syndrome): Decreased levels of estrogen or testosterone occur with gonadal failure. Through a feedback mechanism, FSH and LH secretion is stimulated maximally.
Precocious puberty: One cause of precocious puberty is oversecretion of FSH and LH.
Pituitary adenoma: Some pituitary adenomas secrete FSH or LH without regard to any feedback mechanism.
Decreased Levels
Pituitary failure: FSH and LH are produced in the anterior pituitary. The first indication of pituitary failure is reduction of FSH/LH and the resulting gonadal failure.
Hypothalamic failure: GNRH is produced in the hypothalamus and stimulates FSH/LH production. Failure of that portion of the brain to produce GNRH will cause reduced FSH/LH levels.
Lyme Disease
Test Explanation
Lyme disease was first recognized in Lyme, Connecticut in 1975. It is caused by a spirochete called Borrelia burgdorferi. This is the most common tick-borne disease. The spirochete is spread by a bite from a black-legged tick (Ixodes pacificus) or deer tick (Ixodes scapularis).
Clinical presentation of Lyme disease can either be localized or disseminated. Characteristic of early localized disease is the presence of erythema migrans, a round or oval erythematous skin lesion with a bull's-eye pattern that develops at the site of the tick bite; it is usually present 7 to 14 days after the tick bite and should be ≥5 cm in largest diameter for a firm Lyme disease diagnosis. Disseminated disease that may affect the musculoskeletal, cardiac, or nervous system can follow erythema chronicum migrans (ECM) within days or weeks and is considered early-stage disseminated disease. Meningoencephalitis, cranial or peripheral neuropathies, myocarditis, atrioventricular nodal block, or arthritis are some of the inflammatory changes that may occur. Lyme carditis may overlap temporally with neurologic Lyme disease (late-stage disseminated disease).
Cultures of the ECM can isolate the spirochete in half of the cases. However, it is difficult to culture and takes a long time to grow. Cultures of the blood or CSF are even less helpful. Currently screening serologic studies are performed for the detection of Lyme disease. Enzyme-linked immunosorbent assay (EIA) is the best diagnostic test for Lyme disease. This test determines titers of specific IgM and specific IgG antibodies to the B. burgdorferi spirochete. Levels of specific IgM antibody peak during the third to sixth week after disease onset and then gradually decline. Titers of specific IgG antibodies are generally low during the first several weeks of illness, reach maximal levels in 4 to 6 months, and often remain elevated for years.
Lyme disease can be confused with various viral infections. In these patients a single titer of specific IgM antibody may suggest the correct diagnosis. Acute and convalescent sera can be tested to verify the diagnosis. The Food and Drug Administration (FDA) recommends that all samples with positive or equivocal results in the Borrelia burgdorferi antibody EIA (screening) should be tested by Western blot. Positive or equivocal EIA screening test results should not be interpreted as truly positive until verified with a confirmatory Western blot assay. The Western blot antibody assay can identify specifically the IgG or the IgM antibody. The Western blot assay is considered positive for IgG if five or more of the 10 significant electrophoretic bands are considered positive for Borrelia burgdorferi specific IgG antibody. The Western blot IgM antibody assay is considered positive if two or more out of three significant electrophoretic bands are considered positive for Borrelia burgdorferi IgM antibody. However, the screening test and/or Western blot for B. burgdorferi antibodies may be falsely negative in early stages of Lyme disease, including the period when erythema migrans is apparent.
It is important to note that the diagnosis of Lyme disease can be made with certainty only when the clinical picture of the acute disease and the serologic results both support the diagnosis. Without the clinical picture, serologic tests are often falsely positive and the diagnosis is incorrectly made. The Centers for Disease Control and Prevention (CDC) requires the following for the diagnosis to be made with certainty:
• Isolation of B. burgdorferi from an infected tissue or specimen
• Identification of IgM and IgG antibodies to B. burgdorferi in the blood or CSF
• Acute and convalescent blood samples with significant positive antibody titers
Patients with suspected Lyme disease should have the serologic test repeated if the initial test is negative. Amplification of Borrelia genomic DNA by real-time PCR testing can be performed on cerebrospinal fluid or urine to support the diagnosis. Ticks, after about 36 hours of attachment, may be tested by molecular methods to identify B. burgdorferi.
Magnesium (Mg)
Test Explanation
Most of the magnesium is found in the body intracellularly. About half is in the bone. Most of the magnesium is bound to an adenosine triphosphatase (ATP) molecule and is important in phosphorylation of ATP (main source of energy for the body). Therefore this electrolyte is critical in nearly all metabolic processes. Furthermore, magnesium acts as a cofactor that modifies the activity of many enzymes. Carbohydrate, protein, and nucleic acid synthesis and metabolism depend on magnesium.
Most organ functions, including neuromuscular tissue, also depend on magnesium. It is important to monitor magnesium levels in cardiac patients. Low magnesium levels may increase cardiac irritability and aggravate cardiac arrhythmias. Hypermagnesemia retards neuromuscular conduction and is demonstrated as cardiac conduction slowing (widened PR and Q-T intervals with wide QRS), diminished deep-tendon reflexes, and respiratory depression.
As intracellular elements, body levels of potassium, magnesium, and calcium (in order of quantity) are closely linked. The intracellular electrical charge must be maintained. When the level of one of these positive electrically charged elements is low, another positively charged element is driven into the intracellular space to maintain electrical neutrality. Extracellular and blood levels therefore decrease. A total body reduction in one of those elements creates a comparable blood reduction in the others. Magnesium is closely related to calcium in that it increases the intestinal absorption of calcium. Magnesium is also important in calcium metabolism. Often hypocalcemia will respond to magnesium replacement.
Magnesium deficiency occurs in patients who are malnourished because of malabsorption or maldigestion or lack of food intake. This becomes especially significant in postoperative patients, who may not eat for 5 to 7 days and whose metabolism (and therefore the need for magnesium) is accelerated. Alcohol abuse increases magnesium loss in the urine. Moderate hypomagnesemia occurs with diabetes, hypoparathyroidism, hyperthyroidism, and hyperaldosteronism. Toxemia of pregnancy is also believed to be associated with reduced magnesium levels. Symptoms of magnesium depletion are mostly neuromuscular (i.e., weakness, irritability, tetany, EKG changes, delirium, and convulsions).
Increased magnesium levels most commonly are associated with ingestion of magnesium-containing antacids. Most of the serum magnesium is excreted by the kidney; therefore chronic renal diseases also cause elevated magnesium levels. Several drug interactions also can result in decreased or increased magnesium levels. Because magnesium is an intracellular cation, hemolysis of the collected blood sample should be avoided. Hemolysis will create falsely elevated levels of magnesium. Symptoms of increased magnesium levels include lethargy, nausea and vomiting, and slurred speech.
Interfering Factors
• Hemolysis should be avoided when collecting this specimen. Magnesium is an intracellular ion, and lysis of red blood cells (RBCs) will release great quantities of magnesium into the blood and cause falsely high results.
Drugs that increase magnesium levels include antacids, aminoglycoside antibiotics, calcium-containing medication, laxatives, lithium, loop diuretics, and thyroid medication.
Drugs that decrease magnesium levels include some antibiotics, diuretics, and insulin.
Test Results and Clinical Significance
Increased Levels
Renal insufficiency: Magnesium is excreted by the kidneys. With end-stage renal failure, excretion is reduced and magnesium accumulates in the blood. See discussion below regarding decreased magnesium levels in tubular diseases of the kidney.
Addison disease: Aldosterone enhances magnesium excretion. With reduced aldosterone, magnesium excretion is diminished.
Ingestion of magnesium-containing antacids or salts: Magnesium is absorbed from the intestines. Blood levels rise.
Hypothyroidism: The pathophysiology of this observation is not clear.
Decreased Levels
The major source of magnesium is dietary intake and absorption from the intestines. When either is inhibited, magnesium levels in the blood fall. In malabsorption, all fat-soluble vitamins are lost. Vitamin D levels diminish, and hypocalcemia follows. Magnesium levels therefore fall in light of the low calcium (see above).
Hypoparathyroidism: In this disease, calcium levels are reduced. Calcium enhances intestinal absorption of magnesium, and with low calcium levels, magnesium is not well absorbed, so blood levels diminish. In hyperparathyroidism, calcium levels are high and magnesium levels increase.
Alcoholism: Ethanol increases magnesium losses in the urine.
Chronic renal tubular disease: Magnesium is reabsorbed in the renal tubule. Diseases affecting this area of the kidney (e.g., tubular necrosis) or drugs that are toxic to the renal tubule (e.g., aminoglycosides) will allow increased losses of magnesium in the urine.
Diabetic acidosis: With treatment of this disease, magnesium levels fall. As insulin is given to these patients to drive glucose into the cells, magnesium follows and blood levels drop.
Maternal Screen Testing (Maternal Triple Screen, Maternal Quadruple Screen)
Indications
This is a series of tests that are provided to pregnant women in early pregnancy as a screening test to identify potential birth defects or serious chromosomal/genetic abnormalities.
Test Explanation
These screening tests may indicate the potential for the presence of fetal defects (particularly trisomy 21 [Down syndrome] or trisomy 18). They may also indicate increased risk for neural tube defects (e.g., myelomeningocele, spina bifida) or abdominal wall defects (omphalocele or gastroschisis).
The incidence of these abnormalities is directly related to maternal age. In the United States maternal screening is routinely offered to all pregnant women, usually in their second trimester of pregnancy. Patients must understand that this is a screening test, not a diagnostic test. If the screening tests are positive, more accurate definitive testing, such as chorionic villus sampling (CVS) in early pregnancy or amniocentesis in mid-pregnancy, is recommended. Most pregnant women greater than 35 years of age routinely have CVS or amniocentesis without maternal screening. FISH CVS or amniotic fluid testing (see Laboratory Genetics, p. 1104) for aneuploidy provides rapid detection of chromosome abnormalities.
Several variations of this test are available:
• Double test: Measures two markers, hCG (p. 304) and alpha-fetoprotein (AFP, p. 54).
• Triple test (maternal triple screen test): Measures three markers, human chorionic gonadotropic (hCG), AFP, and estriol (p. 226) AFP is produced in the yolk sac and fetal liver. Unconjugated estriol and hCG are produced by the placenta.
• Quadruple test: Measures four markers, hCG, AFP, estriol, and inhibin A.
• Fully integrated screen test: Measures AFP, estriol, fetal nuchal translucency (p. 888), beta and total hCG, and pregnancy-associated plasma protein-A (PAPP-A, p. 414).
The maternal triple screen test offers a 50% to 80% chance of detecting pregnancies with trisomy 21 as compared with AFP alone, which has only a 30% chance of detection. The Quadruple screen is now routinely recommended and is combined with fetal nuchal translucency [FNT] (see Pelvic Ultrasonography, p. 887). These tests are most accurately performed during the second trimester of pregnancy, more specifically between the 14th and 24th weeks (ideal 16-18 weeks). The use of ultrasound to accurately indicate gestational age improves the sensitivity and specificity of maternal serum screening.
First trimester screening for genetic defects is an option for pregnant women. This testing would include FNT combined with the beta subunit of hCG (beta-hCG, p. 304), and pregnancy-associated plasma protein-A (PAPP-A, p. 414). A low level of PAPP-A may indicate an increased risk for having a stillborn baby. These tests have detection rates comparable to standard second-trimester triple screening.
First trimester (11-13 weeks) screening offers several potential advantages over second-trimester screening. When test results are negative, it may help reduce maternal anxiety earlier. If results are positive, it allows women to take advantage of first trimester prenatal diagnosis by CVS at 10 to 12 weeks or early pregnancy amniocentesis. Detecting problems earlier in the pregnancy may allow women to prepare for a child with health problems. It also affords women greater privacy and less health risk if they elect to terminate the pregnancy. In first trimester testing, open neural tube defects cannot be determined
With trisomy 21, second trimester absolute maternal serum levels of AFP and unconjugated estriol are about 25% lower than normal levels and maternal serum hCG is approximately two times higher than the normal hCG level. The results of the screening are expressed in multiples of median (MoM). AFP and urinary estriol (E3) values during pregnancies with trisomy 21 are lower than those associated with normal pregnancies, which means that values below the mean are below 1 MoM. The hCG value for trisomy 21 is greater than 1 MoM. The MoM, fetal age, and maternal weight are used to calculate the possible risk for chromosomal abnormalities (e.g., trisomy 21). All of the previously named maternal screening tests are discussed elsewhere in this book. For the sake of thoroughness, Inhibin A is discussed here.
Inhibin A is normally secreted by the granulosa cells in the ovaries and inhibits the production of follicle-stimulating hormone (FSH) by the pituitary gland. Inhibin A is a glycoprotein of placental origin in pregnancy similar to hCG. Levels in maternal serum remain relatively constant through the 15th to 18th week of pregnancy. Inhibin A is important in the control of fetal development. Maternal serum levels of inhibin A are twice as high in pregnancies affected by trisomy 21 as in unaffected pregnancies. The discovery of this fact led to the inclusion of inhibin A in the serum screening tests for trisomy 21. Inhibin A concentrations are significantly lower in women with normal pregnancies than in women with pregnancies that result in spontaneous abortions. Furthermore circulating concentrations of inhibin A appear to reflect tumor mass for certain forms of ovarian cancer. More accurate diagnostic testing is required if screening tests are abnormal.
Pregnancy-associated plasma protein-A (PAPP-A) is discussed on p. 414.
It is important to recognize that maternal screening provide only an estimation of risk and not a diagnosis. A negative result indicates that the estimated risk falls below the screen cutoff. A positive result indicates that the estimated risk exceeds the screen cutoff. Neither is a diagnosis of normal or abnormal, respectively. Maternal screen can be performed sequentially. The cutoffs of risks differ depending on the timing of testing. For example for Down syndrome, Sequential Maternal Screening, Part 1 (performed in the first trimester), serum results are negative when the calculated risk is less than 1/50 (2%). If Part 1 is negative, an additional specimen is submitted in the second trimester. With Sequential Maternal Screening, Part 2, serum results are negative when the calculated risk is less than 1/270 (0.37%). Negative results mean that the risk is less than the established cutoff; they do not guarantee the absence of Down syndrome. Results are positive when the risk is greater than the established cutoff (i.e., >1/50 in Sequential Maternal Screening, Part 1, and greater than 1/270 in Sequential Maternal Screening, Part 2). Positive results are not diagnostic. When both Sequential Maternal Screening Part 1 and Part 2 are performed with a screen cutoff of 1/270, the combination of maternal age, nuchal translucency (NT), pregnancy-associated plasma protein A (PAPP-A), alpha-fetoprotein (AFP), unconjugated estriol (uE3), human chorionic gonadotropin (hCG), and inhibin A has an overall detection rate of approximately 90% with a false-positive rate of approximately 3% to 4%. In practice, both the detection rate and false-positive rate vary with maternal age. These numbers change when looking at risk for other abnormalities, such as trisomy 18 or neural tube defects.
Related Tests
Alpha-Fetoprotein (p. 54). This test is one of the commonly performed screening tests for possible birth defects.
Pelvic Ultrasonography (p. 887). This test includes a description of fetal nuchal translucency, an accurate screening test for chromosomal abnormalities.
Human Placental Lactogen (p. 308). This is an accurate test to indicate the presence of fetal distress, disease, or growth restriction.
Pregnancy-Associated Plasma Protein-A (p. 414). This is a maternal screening test for potential birth abnormalities.
Metanephrine, Plasma Free
Test Explanation
Pheochromocytomas, although rarely a cause of hypertension, are dangerous tumors that should be investigated. These tumors produce several catecholamines that can cause episodic or persistent hypertension that is unresponsive to treatment. The current diagnosis of pheochromocytoma depends on biochemical evidence of catecholamine production by the tumor. The best test to establish the diagnosis has not been determined.
Until recently, urinary vanillylmandelic acid (VMA) and catecholamine measurements (see p. 975) were used. Urinary testing is not as sensitive as plasma testing. The low prevalence of these tumors among the tested population and the inadequate sensitivity and specificity of urinary testing made diagnosis of pheochromocytoma cumbersome and time-consuming. The development of high-performance liquid chromatography (HPLC) has allowed for more sensitivity in measuring plasma-free metanephrine levels. This is a blood test that measures the amount of metanephrine and normetanephrine, which are metabolites of epinephrine and norepinephrine, two catecholamines.
The high sensitivity of plasma-free metanephrine testing provides a high negative predictive value to the test. This means that if the concentrations of the free metanephrines in the blood are normal, it is very unlikely that a patient has a pheochromocytoma. False positives do occur, though rarely. The diagnostic superiority of plasma metanephrines over plasma or urinary catecholamines and urinary VMA is clear. In about 80% of patients with pheochromocytoma, the magnitude of increase in plasma-free metanephrines is so large that the tumor can be confirmed with close to 100% probability. Intermediate concentrations of normetanephrine and metanephrine are considered indeterminate.
Urinary testing may clarify indeterminate findings. However, comparison of plasma metanephrines and urine metanephrines requires caution because different catecholamine metabolites are measured. Testing for some urinary catecholamines may be more specific than for plasma-free metanephrines, meaning that false positives are less common with urinary testing.
When interpreting results, the following may be helpful:
• Any sample in which the concentrations of both normetanephrine and metanephrine are less than the upper reference range limit should be considered normal, and the presence of pheochromocytoma is highly unlikely.
• Any sample where the concentrations of either normetanephrine or metanephrine exceed their respective upper reference range limits should be considered elevated.
• Whenever the normetanephrine or metanephrine concentration exceeds the indeterminate range, the presence of pheochromocytoma is highly probable and should be located via imaging techniques.
Interfering Factors
• Increased levels of metanephrines may be caused by caffeine or alcohol.
• Vigorous exercise, stress, and starvation may cause increased metanephrine levels.
Drugs that may cause increased metanephrine levels include epinephrine- or norepinephrine-containing drugs, levodopa, lithium, and nitroglycerin.
Acetaminophen can interfere with HPLC testing of metanephrines and should be avoided for 48 hours before testing.
Procedure and Patient Care
During
• Identify and minimize factors contributing to patient stress and anxiety. Physical exertion and emotional stress may alter metanephrine test results.
• The patients may be asked to lie down and rest quietly for 15 to 30 minutes before sample collection.
• The blood sample may be collected while supine.
• Collect a venous blood sample in a chilled lavender-top (EDTA) or pink-top (K2EDTA) tube. Invert to mix with preservatives.
Related Test
Vanillylmandelic Acid and Catecholamines (p. 975). This 24-hour urine test for VMA and catecholamines is performed primarily to diagnose hypertension secondary to pheochromocytoma, neuroblastomas, and other rare adrenal tumors.
Methemoglobin (Hemoglobin M)
Test Explanation
Methemoglobin is continuously formed in the red blood cells (RBCs). During the production of normal adult deoxygenated hemoglobin, methemoglobin is reduced to normal adult deoxygenated hemoglobin by nicotinamide adenine dinucleotide dependent reductase enzyme (NADH). If oxygenation of the iron component in the protohemoglobin occurs without subsequent reduction of the heme iron back to its Fe+2 form as exists in normal hemoglobin, excess methemoglobin accumulates. The oxidized iron form in methemoglobin is unable to combine with oxygen to carry the oxygen to the peripheral tissues. Therefore the oxyhemoglobin dissociation curve is “shifted to the left” resulting in cyanosis and hypoxia. Elderly, pediatric, or chronically hypoxemic patients are particularly sensitive to methemoglobin production.
Methemoglobinemia can be congenital or, more commonly, is acquired. Hemoglobin M disease is a genetic defect that results in a group of abnormal hemoglobins that are methemoglobins. Another genetic mutation can cause a deficiency in NADH methemoglobin reductase enzyme that is required to deoxygenate methemoglobin to normal adult hemoglobin. These forms of methemoglobinemia occur in infants, are usually severe, are not amenable to treatment, and are often fatal.
Acquired methemoglobinemia is a result of ingestion of nitrates (e.g., from well water), or drugs such as phenacetin, sulfonamides, isoniazid, local anesthetics containing benzocaine, sulfonamide antibiotics, silver nitrate, and Pyridium. Several over-the-counter local anesthetics used for toothache or hemorrhoidal pain contain benzocaine. The acquired form of the disease commonly occurs in older individuals and results in an acute crisis that is effectively treated with ascorbic acid or methylene blue. However, methylene blue is contraindicated in G6PD deficiency.
Microglobulin (Beta-2 Microglobulin [B2M], Alpha 1 Microglobulin, and Retinol-Binding Protein)
Indications
This test is used to evaluate patients with malignancies, chronic infections, inflammatory diseases, and renal diseases.
Test Explanation
Beta-2 microglobulin (B2M) is a protein found on the surface of all cells. It is an HLA major histocompatibility antigen that exists in increased numbers on the cell surface and particularly on lymphatic cells. Production of this protein increases with cell turnover. B2M is increased in patients with malignancies (especially B-cell lymphoma, leukemia, or multiple myeloma), chronic infections, and in patients with chronic severe inflammatory diseases. It is an accurate measurement of myeloma tumor disease activity, stage of disease, and prognosis and, as such, is an important tumor marker. This tumor marker is best determined in the blood.
B2M, alpha 1 microglobulin, and retinol-binding proteins pass freely through glomerular membranes and are near completely reabsorbed by renal proximal tubules cells. Because of extensive tubular reabsorption, under normal conditions very little of these proteins appear in the final excreted urine. Therefore an increase in the urinary excretion of these proteins indicates proximal tubule disease or toxicity and/or impaired proximal tubular function. In patients with a urinary tract infection, these proteins indicate pyelonephritis. These proteins are helpful in differentiating glomerular from tubular renal disease. In patients with aminoglycoside toxicity, heavy metal nephrotoxicity, or tubular disease, protein urine levels are elevated. Excretion is increased 100 to 1000 times normal levels in cadmium-exposed workers. This test is used to monitor these workers. Periodic testing is performed on these patients to detect kidney disease at its earliest stage. To date there are no convincing studies to indicate that one protein has better clinical utility than the other.
B2M is particularly helpful in the differential diagnosis of renal disease. If blood and urine levels are obtained simultaneously, one can differentiate glomerular from tubular disease. In glomerular disease, because of poor glomerular filtration, blood levels are high and urine levels are low. In tubular disease, because of poor tubular reabsorption, the blood levels are low and urine levels are high. Blood levels increase early in kidney transplant rejection.
Urinary excretion of these proteins can be determined from either a 24-hour collection or a random urine collection. The 24-hour collection is traditionally considered the gold standard. For random or spot collections, the concentration of alpha-1-microglobulin is divided by the urinary creatinine concentration. This corrected value adjusts alpha-1-microglobulin for variabilities in urine concentration.
Increased CSF levels of B2M indicate central nervous system involvement with leukemia, lymphoma, HIV, or multiple sclerosis.
Quantitative chemiluminescent immunoassay or nephelometry methods are used to identify these proteins in the urine or serum.
Procedure and Patient Care
During
Urine
 See Box 11-2, Guidelines for a 24-Hour Urine Collection, p. 907.
See Box 11-2, Guidelines for a 24-Hour Urine Collection, p. 907.
Encourage the patient to drink fluids during the 24 hours unless this is contraindicated for medical purposes.
• If a single random urine collection is requested, collect specimen for protein and creatinine testing to adjust for urine concentration.
Test Results and Clinical Implications
Increased Urine Levels
Mononucleosis Rapid Test (Mononuclear Heterophil Test, Heterophil Antibody Test, Monospot Test)
Indications
This is a rapid slide test designed to assist in the diagnosis of infectious mononucleosis.
Test Explanation
The mononucleosis rapid test is performed to make the diagnosis of infectious mononucleosis (IM), a disease caused by the Epstein-Barr virus (EBV). Usually young adults are affected by mononucleosis. The clinical presentation is fever, pharyngitis, lymphadenopathy, and splenomegaly. Detectable levels of the IM heterophile antibody can usually be expected to occur between the sixth and tenth day following the onset of symptoms. The level usually increases through the second or third week of illness and, thereafter, can be expected to persist, gradually declining over a 12-month period. The IM heterophile antibody has been associated with several diseases other than IM. These include leukemia, Burkitt lymphoma, pancreatic carcinoma, viral hepatitis, cytomegalovirus infections, and others.
Several heterophil agglutination tests are available, but the most frequently performed is the rapid slide test for infectious mononucleosis (previously called the Monospot test). In most tests, a suspension of polystyrene latex particles is coated with a highly purified antigen from bovine red cell membranes. The degree of purity of the antigen is such that it only reacts with infectious mononucleosis heterophile antibodies. If infectious mononucleosis heterophile antibodies are present in serum, the latex suspension changes its uniform appearance and a clear agglutination becomes evident, indicating an EBV virus infection. EBV immunologic quantification (p. 312) is available when IM is suspected but the Mono Spot is negative.
The diagnosis of infectious mononucleosis must include the following criteria:
Related Test
Epstein-Barr Virus Testing (p. 217). These tests are recommended only when a mononucleosis screening procedure is negative and infectious mononucleosis or a complication of Epstein-Barr virus infection is suspected. Also they more precisely define the acuity of the infection.
Mycoplasma pneumoniae Antibodies, IgG and IgM
Indications
This test is used to support the clinical diagnosis of disease associated with Mycoplasma pneumoniae.
Test Explanation
Several diseases have been associated with the mycoplasmal pneumoniae infection, including pharyngitis, tracheobronchitis, pneumonia, and inflammation of the tympanic membrane. Mycoplasma pneumoniae accounts for approximately 20% of all cases of pneumonia. Classically it causes a disease that has been described as primary atypical pneumonia. The disease is of insidious onset with fever, headache, and malaise for 2 to 4 days before the onset of respiratory symptoms. Most patients do not require hospitalization. Symptomatic infections attributable to this organism most commonly occur in children and young adults. These infections may be associated with cold agglutinin syndrome (p. 170).
Positive IgM results are consistent with acute infection, although there may be some cross-reactivity associated with other mycoplasma infections. A single positive IgG result only indicates previous immunologic exposure. Negative results do not rule out the presence of Mycoplasma pneumoniae–associated disease because the specimen may have been drawn before the appearance of detectable antibodies. If a Mycoplasma infection is clinically suspected, a second specimen should be submitted at least 14 days later. The continued presence or absence of antibodies cannot be used to determine the success or failure of therapy.
After serologic combinations to identify IgG/IgM antibody complexes, serial dilutions are performed and the color changes are measured photometrically. The color intensity of the dilutions depends on the antibody concentration in the serum sample. The IgM complex can be identified by immunofluorescent assay (IFA).
Related Tests
Cold Agglutinins (p. 170). This test identifies antibodies to RBCs that cause agglutination when exposed to cold temperatures. Although other diseases are also associated with the presence of these antibodies, Mycoplasma infections are considered predominant.
Myoglobin
Indications
This test is used in the early evaluation of a patient with suspected acute myocardial infarction (MI). It is also used to assist in the diagnosis of disease or injury of skeletal muscle.
Test Explanation
Myoglobin is an oxygen-binding protein found in cardiac and skeletal muscle. Measurement of myoglobin provides an early index of damage to the myocardium, such as occurs in myocardial infarction (MI) or reinfarction. Increased levels, which indicate cardiac muscle injury or death, occur in about 3 hours. Although this test is more sensitive than creatine phosphokinase (CPK) isoenzymes, it is not as specific. Trauma, inflammation, or ischemic changes to the noncardiac skeletal muscles can also cause elevated levels of myoglobin. The benefit of myoglobin over CPK-MB (see p. 186) is that it may become elevated earlier in some patients. This may prove beneficial because thrombolytic therapy should be started within the first 6 hours after an MI.
As already indicated, disease or trauma of the skeletal muscle also causes elevations in myoglobin. With sudden and severe muscle injury, myoglobin can reach very high levels. Because myoglobin is excreted in the urine and is nephrotoxic, urine levels must be monitored in patients with high levels. To screen for myoglobin, the routine urine dipstick for hemoglobin can be used.
Interfering Factors
• Recent administration of radioactive substances may affect test results determined by radioimmunoassay (RIA) methods. The recently administered radioisotope may interfere with testing.
• Increased myoglobin levels can occur after intramuscular (IM) injections. The injection can cause localized muscle injury and instigate an inflammatory response that could elevate myoglobin levels.
Test Results and Clinical Significance
Increased Levels
MI: Injury to cardiac muscle causes the cells to lyse and expel the myoglobin into the bloodstream.
Skeletal muscle inflammation (myositis): Injury to skeletal muscle causes the cells to lyse and expel the myoglobin into the bloodstream.
All of these diseases affect the skeletal muscles. This causes the muscle cells to lyse and expel the myoglobin into the bloodstream.
Seizures: Persistent seizure activity injures skeletal muscle tissue. This causes the muscle cells to lyse and expel the myoglobin into the bloodstream.
Natriuretic Peptides (Atrial Natriuretic Peptide [ANP], Brain Natriuretic Peptide [BNP], C-type Natriuretic Peptide [CNP], B-Type Natriuretic Peptide [BNP], Ventricular Natriuretic Peptide, CHF Peptides)
Indications
Natriuretic peptides are used to identify and stratify patients with congestive heart failure (CHF).
Test Explanation and Related Physiology
Natriuretic peptides (NPs) are used to identify and stratify patients with congestive heart failure (CHF). NPs are neuroendocrine peptides that oppose the activity of the renin-angiotensin system. There are three major NPs: ANP, BNP, and CNP. ANP is synthesized in the cardiac atrial muscle. The main source of BNP is the membrane granules in the cardiac ventricle, although it was initially found in porcine brain. CNP was first localized in the nervous system but later found to be produced by the endothelial cells.The cardiac peptides are continuously released by the heart muscle cells in low levels. But, the rate of release can be increased by a variety of neuroendocrine and physiologic factors, including hemodynamic load, to regulate cardiac preload and afterload. Because of these properties, BNP and ANP have been implicated in the pathophysiology of hypertension, CHF, and atherosclerosis. Both ANP and BNP are released in response to atrial and ventricular stretch, respectively, and will cause vasorelaxation, inhibition of aldosterone secretion from the adrenal gland and renin from the kidney, thereby increasing natriuresis and reduction in blood volume. CNP has a vasorelaxation effect but does not stimulate natriuresis.
BNP, in particular, correlates well to left ventricular pressures. As a result, BNP is a good marker for CHF. BNP levels, by themselves, are more accurate than any historical or physical findings or laboratory values in identifying congestive heart failure as the cause of dyspnea. The diagnostic accuracy of BNP at a cutoff of 100 pg/mL was 83.4% in research studies.
The higher the levels of BNP are, the more severe the CHF. This test is used in urgent care settings to aid in the differential diagnosis of shortness of breath (SOB). If BNP is elevated, the SOB is because of CHF. If BNP levels are normal, the SOB is pulmonary and not cardiac. This is particularly helpful in evaluating SOB in patients with cardiac and chronic lung disease.
Furthermore, BNP is a helpful prognosticator and is used in CHF risk stratification. CHF patients whose BNP levels do not rapidly return to normal with treatment experience a significantly higher risk for mortality in the ensuing months than do those whose BNP levels rapidly normalize with treatment. In early rejection of heart transplants, BNP levels can be elevated. Measurement of plasma BNP concentration is evolving as a very efficient and cost effective mass screening technique for identifying patients with various cardiac abnormalities. This measurement is important regardless of etiology and degree of left ventricular (LV) systolic dysfunction that can potentially develop into obvious heart failure and carry a high risk for a cardiovascular event.
In some laboratories, BNP is measured as an N-terminal fragment of pro–brain (B-type) natriuretic peptide (NT–pro-BNP). The clinical information provided by either the BNP or the pro-BNP is about the same and the tests are used interchangeably. In most clinical laboratories, BNP is performed by automated systems using chemiluminescence (e.g., Beckman Coulter). Screening diabetics for BNP elevation to determine the risk for cardiac diseases is used because of the low cost of performing the test as compared with an echocardiogram. BNP is also elevated in patients with prolonged systemic hypertension and those with acute myocardial infarction (MI).
Interfering Factors
• BNP levels are generally higher in healthy women than healthy men.
• BNP levels are higher in older patients.
• BNP levels are elevated in patients who have had cardiac surgery for 1 month postoperatively. This does not reflect the presence of CHF.
• There are several different methods of measuring BNP. Normal values vary whether or not the whole protein of a BNP fragment protein is measured.
Natrecor (nesiritide), a recombinant form of the endogenous human peptide used to treat CHF, will increase plasma BNP levels for several days.
Test Results and Clinical Significance
Increased Levels
These diseases are all associated with increased ventricular and/or atrial cardiac pressure. As a result, cardiac natriuretic peptides are secreted, causing a relaxation of blood vessels (vasodilation), an increase in the excretion of sodium (natriuresis) and fluid (diuresis), and a decrease in injurious neurohormones (endothelin, aldosterone, angiotensin II). All of these actions work in concert on the vessels, heart, and kidney to decrease the fluid load on the heart, allowing the heart to function better and improving cardiac performance.
Related Test
Chest X-Ray (p. 1014). This test provides information about the heart, lungs, bony thorax, mediastinum, and great vessels.
Neuron-Specific Enolase (NSE)
Indications
This test is used as a marker in patients with neuron-specific enolase-secreting tumors (e.g., carcinoids, small cell lung carcinoma, neuroblastomas). It is also used as an auxiliary tool in the assessment of comatose patients: the higher the NSE level, the more injury to the central nervous system.
Test Explanation
NSE is a glycolytic enzyme that catalyzes the conversion of phosphoglycerate to phosphoenol pyruvate. It is present in neuronal, neuroendocrine, and amine precursor uptake decarboxylation (APUD) cells. NSE, in serum or cerebrospinal fluid (CSF), is often elevated in diseases, which result in neuronal destruction. Measurement of NSE in serum of CSF therefore can assist in the differential diagnosis of a variety of neuron-destructive and neurodegenerative disorders. NSE might also have utility as a prognostic marker in neuronal hypoxic injury.
Elevated NSE concentrations are observed in patients with neuroblastoma, pancreatic islet cell carcinoma, medullary thyroid carcinoma, pheochromocytoma, and other neural crest–derived or neuroendocrine tumors. NSE levels are frequently increased in patients with small cell lung cancer (SCLC) and infrequently in patients with non-SCLC. When increased, NSE can be used to monitor disease progression and management in SCLC. Levels of NSE occasionally can be elevated in benign disorders, such as pneumonia and benign hepatobiliary diseases.
NSE values can vary significantly among methods and assays. Serial follow-up should be performed with the same assay. If assays are changed, patients should have new baseline values. Immunometric assays are most commonly used.
Test Results and Clinical Significance
Related Test
Magnetic Resonance Imaging (p. 1106). This noninvasive diagnostic procedure provides invaluable information about the brain, central nervous system, bones, joints, breasts, and so on.
Neutrophil Antibody Screen (Granulocyte Antibodies, Polymorphonucleocyte Antibodies [PMN ab], Antigranulocyte Antibodies, Antineutrophil Antibodies, Neutrophil Antibodies)
Indications
This test is performed to identify antibodies to white blood cells (WBCs) if blood transfusion is associated with an immune reaction.
Test Explanation
Neutrophil antibodies are directed toward WBCs. They develop during blood transfusions. Patients who experience a transfusion reaction despite complete compatibility testing before blood administration should have a neutrophil antibody screen to see if WBC incompatibility is the source of the reaction. This test is most commonly a part of post-transfusion antibody screening, which is a battery of testing performed if a transfusion reaction is suspected (see Boxes 2-15 and 2-16).
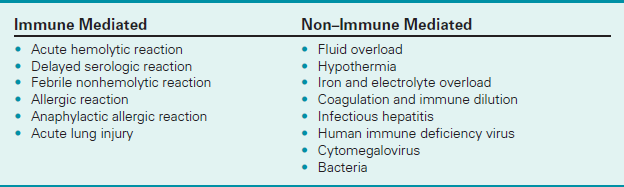
Most commonly, the recipient has antibodies to the donor WBCs and will experience a fever during transfusion. More severe, however, is the reaction when the donor plasma contains antibodies to the recipient's WBCs. This nonhemolytic reaction can lead to severe transfusion reactions, including acute pulmonary failure (transfusion-related acute lung injury [TRALI]) and multiorgan system failure. The majority of TRALI cases can be triggered by passive transfer of human lymphocyte antigen (HLA) or neutrophil-specific antibodies from the donor to the recipient. TRALI is the leading cause of transfusion-related mortality rates and accounts for 13% of all transfusion deaths.
Related Tests
Coombs Test, Direct (p. 175). This test is performed to identify hemolysis (lysis of red blood cells [RBCs]) or to investigate hemolytic transfusion.
Coombs Test, Indirect (p. 177). This test is used to detect antibodies against RBCs in the serum. It is most commonly used for screening potential blood recipients.
Neutrophil Gelatinase–Associated Lipocalin (NGAL, Lipocalin-2)
Indications
NGAL is a predictor for acute kidney injury (AKI), previously referred to as acute renal failure, and chronic kidney disease (CKD).
Test Explanation
There are no early markers for acute or chronic renal disease. Serum creatinine levels rise only after there has been significant renal impairment and injury. It is important to note that the earlier renal disease or injury is identified, the more successfully it can be treated. Early treatment also helps to lower the morbidity associated with the disease. This is particularly important in patients who have serious nonrenal disease (e.g., heart surgery, renal transplant, sepsis). In these patients, severe acute kidney injury (AKI) increases morbidity and mortality of hospitalized patients.
NGAL is a member of the lipocalin family of proteins, which bind and transport small lipophilic molecules. NGAL is generally expressed in low concentrations from the renal tubules, but it increases greatly in the presence of epithelial injury and inflammation. A marked elevation in NGAL indicates that renal injury has occurred and aggressive supportive treatment should be instituted. NGAL concentrations rise 48 hours before a rise in creatinine is noted. NGAL can be detected in both urine and blood within two hours of a renal insult.
NGAL can be measured in the urine, plasma, or serum samples with ELISA test kits. Results are available in less than 1 hour in a standard laboratory with conventional ELISA equipment. This is particularly helpful in an intensive care environment. By itself, the absolute baseline laboratory result is not as important as are the succeeding results. Normal values vary according to which laboratory method is used and the patient's baseline GFR. NGAL varies inversely with the GFR. Urine or blood samples can also be analyzed using an established and validated enzyme immunoassay (EIA).
NGAL measurements are being used increasingly in a variety of clinical situations leading to AKI (such as during cardiac surgery, kidney transplantation, contrast nephropathy, and hemolytic uremic syndrome, and in the intensive care setting). It is also useful in conditions leading to CKD (such as lupus nephritis, glomerulonephritis, obstruction, dysplasia, polycystic kidney disease, IgA nephropathy, renal dysplasia, obstructive uropathy, and glomerular and cystic diseases).
Newborn Metabolic Screening
Indications
Newborn metabolic screening is the practice of testing every newborn for certain harmful or potentially fatal disorders that are not otherwise apparent at birth.
Test Explanation
Newborn screening tests take place before the newborn leaves the hospital. Babies are tested to identify serious or life-threatening (and for the most part preventable or treatable) diseases before symptoms begin. These diseases are usually rare. However, if they are not accurately diagnosed and treated, they can cause mental retardation, severe illness, and premature death in newborns. Many of these are metabolic disorders, often called “inborn errors of metabolism.” Other disorders that may be detected through screening are endocrine or hematologic. In most states, this testing is mandatory.
Within 48 hours of a child's birth, a sample of blood is obtained from a “heel stick,” and the blood is analyzed. The sample, called a “blood spot,” is tested at a reference laboratory. It is generally recommended that the sample be taken after the first 24 hours of life. Some tests, such as the one for phenylketonuria (PKU), may not be as sensitive until the newborn has ingested an ample amount of the amino acid phenylalanine, which is a constituent of both human and cow's milk, and after the postnatal thyroid surge has subsided. This is generally after about 2 days.
With the use of Tandem mass spectrometry (tandem MS), multiple blood tests can be performed quickly and efficiently. When directed to newborn blood screening, the use of these specialized instruments can detect abnormally elevated proteins associated with certain metabolic disorders. They are capable of screening for more than 20 inherited metabolic disorders with a single test in only a few minutes. Tandem MS is very accurate and can measure very small amounts of similar material with excellent precision. For example, PKU (see below) tandem MS has been shown to reduce the false-positive rate (false alarms) for this disorder more than tenfold compared to the best alternative method available. The disorders listed below are the ones typically included in newborn screening programs:
PKU: An inherited disease, PKU is characterized by deficiency of the enzyme phenylalanine hydroxylase, which converts phenylalanine to tyrosine. Phenylalanine is an essential amino acid necessary for growth; however, any excess must be degraded by conversion to tyrosine. An infant with PKU lacks the ability to make this necessary conversion. Thus phenylalanine accumulates in the body and spills over into the urine. If the amount of phenylalanine is not restricted in infants with PKU, progressive mental retardation results. A low-phenylalanine diet will need to be followed throughout childhood and adolescence and perhaps into adult life. (Incidence: 1 in 10,000 to 25,000.)
Congenital hypothyroidism: Affected babies without treatment experience retarded growth and brain development. If the disorder is detected early, a baby can be treated with oral doses of thyroid hormone to permit normal development. (Incidence: 1 in 4000.)
Galactosemia: Babies with galactosemia lack the enzyme that converts galactose into glucose, a sugar the body is able to use. As a result, milk and other dairy products must be eliminated from the diet. Otherwise, galactose can build up and cause blindness, severe mental retardation, growth deficiency, and even death. (Incidence: 1 in 60,000 to 80,000.) There are several less severe forms of galactosemia that may be detected by newborn screening. These may not require any intervention.
Sickle cell anemia: Sickle cell disease is an inherited blood disease in which red blood cells stretch into abnormal “sickle” shapes (see p. 464). This can cause episodes of pain, damage to vital organs (such as the lungs and kidneys), and even death. Young children with sickle cell anemia are especially prone to certain dangerous bacterial infections. The screening test can also detect other disorders affecting hemoglobin (the oxygen-carrying substance in the blood). (Incidence: about 1 in every 500 African American births and 1 in every 1000 to 1400 Hispanic American births.)
Biotinidase deficiency: Babies with this condition do not have enough biotinidase, an enzyme that recycles biotin (one of the B vitamins) in the body. This deficiency may cause seizures, poor muscle control, immune system impairment, hearing loss, mental retardation, coma, and even death. If the deficiency is detected early, however, problems can be prevented by biotin administration. (Incidence: 1 in 126,000.)
Congenital adrenal hyperplasia: This is actually a group of disorders resulting in a deficiency of adrenal hormones. It can affect the development of the genitals and may cause death. Lifelong treatment through hormone supplementation manages the condition. (Incidence: 1 in 12,000.)
Maple syrup urine disease (MSUD): Babies with MSUD are missing an enzyme needed to process the amino acids leucine, isoleucine, and valine (present in protein-rich foods such as milk, meat, and eggs) that are essential for the body's normal growth. When these are not processed properly, they can build up in the body, causing urine to smell like maple syrup or sweet, burnt sugar. These babies usually have little appetite and are extremely irritable. If not detected and treated early, MSUD can cause mental retardation, physical disability, and even death. A carefully controlled diet free of high-protein foods can prevent these outcomes. (Incidence: 1 in 250,000.)
Homocystinuria: This metabolic disorder results from a deficiency in cystathionine β-synthase, responsible for the metabolism of methionine and homocysteine. If untreated, it can lead to dislocated lenses of the eyes, mental retardation, skeletal abnormalities, and hypercoagulability. However, a special diet combined with dietary supplements may help prevent most of these problems. (Incidence: 1 in 50,000 to 150,000.)
Tyrosinemia: Babies with this disorder cannot metabolize tyrosine. If it accumulates in the body, it can cause mild retardation, language skill difficulties, liver problems, and even death from liver failure. A special diet and sometimes a liver transplant are needed to treat the condition. Early diagnosis and treatment seem to offset long-term problems. (Incidence: not yet determined.)
Cystic fibrosis: This is an inherited disorder expressed in the lung and gastrointestinal tract that causes cells to release thick mucus leading to chronic respiratory disease, problems with digestion, and poor growth. There is no known cure; treatment involves trying to prevent the serious lung infections associated with it and providing adequate nutrition. (Incidence: 1 in 2000 white babies.)
Toxoplasmosis: Toxoplasmosis is a parasitic infection that can be transmitted through the mother's placenta to an unborn child. The disease-causing organism, which is found in undercooked meat, can invade the brain, eye, and muscle, possibly resulting in blindness and mental retardation. (Incidence: 1 in 1000.)
These are not the only metabolic disorders that can be detected through newborn screening. Certain other rare disorders can also be detected and include Duchenne's muscular dystrophy, human immune deficiency virus (HIV) infection, and neuroblastoma. Hematologic disorders, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency and thalassemia, can also be identified.
Most, but not all, states require newborns' hearing to be screened before they are discharged from the hospital. The hearing test involves placing a tiny earphone in the baby's ear and measuring his or her response to sound. A child develops critical speaking and language skills in the first few years of life, and if a hearing loss is discovered early, developmental effects on language skills can be avoided.
5'-Nucleotidase
Indications
5'-Nucleotidase is used to support the diagnosis of hepatobiliary obstructive disease. It is especially useful in helping confirm that an elevated alkaline phosphatase (ALP) is the result of liver pathology rather than pathology of another tissue origin.
Test Explanation
5'-Nucleotidase is an enzyme specific to the liver. The 5'-nucleotidase level is elevated in patients with liver diseases, especially those associated with cholestasis. It provides information similar to ALP. However, ALP is not specific to the liver. Diseases of the bone, sepsis, pregnancy, and other disease can produce ALP elevation. When there is doubt regarding the cause of an elevated ALP, a 5'-nucleotidase test is recommended. If that enzyme is elevated along with the ALP, the pathologic source is certainly the liver. If the 5'-nucleotidase is normal in the face of an elevated ALP, the pathologic source is outside the liver (bone, kidney, spleen). Gamma-glutamyl transpeptidase (GGTP) is used similarly, as it is also specific to the liver. GGTP and ALP can also be elevated from drug-induced cholestasis; 5'-nucleotidase cannot.
Osmolality, Blood (Serum Osmolality)
Indications
This test is used to gain information about fluid status and electrolyte imbalance. It is also helpful in evaluating illnesses involving antidiuretic hormone (ADH).
Test Explanation
Osmolality measures the number of dissolved particles in serum/plasma per unit volume. As the amount of free water in the blood increases or the number of particles decreases per unit volume of serum, osmolality decreases. As the amount of water in the blood decreases or the number of particles per unit volume increases, osmolality increases. Osmolality increases with dehydration and decreases with overhydration.
There is an elaborate feedback mechanism that controls osmolality. Increased osmolality will stimulate secretion of ADH. This will result in increased water reabsorption in the kidney, more concentrated urine, and less concentrated serum. A low serum osmolality will suppress the release of ADH, resulting in decreased water reabsorption and large amounts of dilute urine. The simultaneous use of urine osmolality (p. 938) helps in the interpretation and evaluation of problems involving osmolality.
The serum osmolality test is useful in evaluating fluid and electrolyte imbalance. The test is very helpful in the evaluation of seizures, ascites, hydration status, acid-base balance, suspected antidiuretic hormone (ADH) abnormalities, and suspected poisoning. Osmolality is also helpful in identifying the presence of organic acids, sugars, and ethanol.
The osmolality can be predicted based on calculations of serum sodium, glucose, and BUN—the three most important solutes in the blood.
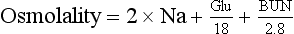
The normal range of serum osmolality is 285 to 295 mOsm/L. The measured osmolality should not exceed the predicted by more than 10 mOsm/L. A difference of more than 10 mOsm/L is considered an osmolal gap or delta gap. Causes for a serum osmolal gap include mannitol, ethanol, methanol, ethylene glycol, and other toxins in very high concentration, usually small molecules. Another measure providing similar data is the ratio of serum sodium to osmolality. Normally the ratio of serum sodium, in mEq/L, to serum osmolality, in mOsm/kg, is between 0.43 and 0.5. The ratio may be distorted in drug intoxication.
Osmolality may have a role in evaluation of coma patients. Values greater than 385 mOsm/kg H2O are associated with stupor in patients with hyperglycemia. When values of 400 to 420 are detected, grand mal seizures can occur. Values greater than 420 can be lethal. The simultaneous use of urine osmolality (p. 938) helps in the interpretation and evaluation of problems with osmolality.
Test Results and Clinical Significance
Increased Levels
Hyperosmolar nonketotic hyperglycemia,
All of the above illnesses are associated with an increase in the number of particles dissolved in the blood.
Dehydration: Decreased replacement of ongoing water losses leads to dehydration and a rise in the serum osmolality.
Decreased Levels
Overhydration: The provision of free water above the ongoing losses creates a situation in which there is excess free water in the blood. Serum osmolality decreases.
Syndrome of inappropriate ADH (SIADH) secretion: Several illnesses can create this syndrome. ADH is inappropriately secreted despite factors that normally would inhibit its secretion. As a result, large quantities of water are reabsorbed by the kidney. The serum becomes dilute and osmolality decreases.
Paraneoplastic syndromes associated with carcinoma (lung, breast, colon): These cancers act as an autonomous ectopic source for the secretion of ADH. The pathophysiology is the same as described above for SIADH.
Related Tests
Urine Osmolality (p. 938). This is a measurement of the concentration of dissolved particles in the urine. When measured with the serum osmolality, greater insight is obtained about problems of fluid balance.
Antidiuretic Hormone (ADH) (p. 73). This is a direct measurement of ADH in the blood.
Antidiuretic Hormone (ADH) Suppression (p. 76). This test is very helpful in the evaluation of ADH abnormalities.
Parathyroid Hormone (PTH, Parathormone)

Indications
PTH is measured to assist in the evaluation of hypercalcemia or hypocalcemia. It is routinely monitored in patients with chronic renal failure (CRF).
Test Explanation
PTH is the only hormone secreted by the parathyroid gland in response to hypocalcemia. When calcium serum levels return to normal, PTH levels diminish. PTH, therefore, is one of the major factors affecting calcium metabolism. This test is useful in establishing a diagnosis of hyperparathyroidism and distinguishing nonparathyroid from parathyroid causes of hypercalcemia. Increased PTH levels are seen in patients with hyperparathyroidism (primary, secondary, or tertiary); in patients with nonparathyroid, ectopic PTH-producing tumors (pseudohyperparathyroidism); or as a normal compensatory response to hypocalcemia in patients with malabsorption or vitamin D deficiency.
Primary hyperparathyroidism is most often caused by a parathyroid adenoma and only rarely from parathyroid cancer. These patients have high PTH and calcium levels. Secondary hyperparathyroidism is the exaggerated response of the parathyroid gland to kidney insensitivity to PTH in CRF patients. CRF patients have chronically low serum calcium levels in reaction to persistently high levels of phosphate that the kidney fails to excrete. In response to this persistently low calcium, the parathyroid is constantly stimulated to produce PTH to attempt to maintain a normal calcium level. This is called secondary hyperparathyroidism. These patients have high PTH and normal to slightly low calcium levels. Occasionally a patient with CRF overshoots the compensatory process and autonomously develops unnecessarily high PTH production that leads to hypercalcemia. This is called tertiary hyperparathyroidism. These patients have high PTH and high calcium levels.
It is important to measure serum calcium (see p. 138) simultaneously with PTH. Most laboratories have a PTH/calcium nomogram already made up indicating what PTH level is considered normal for each calcium level.
Decreased PTH levels are seen in patients with hypoparathyroidism or as a compensatory response to hypercalcemia in patients with metastatic bone tumors, sarcoidosis, vitamin D intoxication, or milk-alkali syndrome. Of course, surgical ablation of the parathyroid glands is another cause of hypoparathyroidism.
Whole (intact) PTH is metabolized to several different fragments, including an amino or N-terminal, a midregion or midmolecule, and a carboxyl or C-terminal. The intact PTH and the N-terminal are metabolically active. These can all be measured by immunoassay. The intact PTH and all fragments generally provide accurate information concerning the level of PTH in the blood. The intact PTH is probably most often tested as it is most reliable.
Interfering Factors
• Recent injection of radioisotopes may interfere with this test if radioimmunoassay (RIA) methods are used. However, RIA is not a frequently used method of measuring PTH fragments.
Drugs that increase PTH include anticonvulsants, isoniazid, lithium, phosphates, rifampin, and steroids.
Drugs that decrease PTH include cimetidine, pindolol, and propranolol.
Clinical Priorities
• This test is routinely monitored in patients with CRF. These patients have chronically low serum calcium levels in reaction to persistently high phosphate levels that the kidney fails to excrete.
• It is important to measure serum PTH and serum calcium levels at the same time. These values are important for a differential diagnosis. Most laboratories have PTH/calcium nomograms indicating normal PTH levels for each calcium level.
• PTH levels are affected by a diurnal variation. Levels are highest around 2 AM and lowest around 2 PM. Usually an 8 AM blood specimen is drawn. If the patient works nights, the laboratory should be notified so that changes in the diurnal variation can be factored in.
Procedure and Patient Care
During
• Obtain an 8 AM blood specimen, because diurnal rhythm affects PTH levels. (Check with the laboratory if the patient works at night.) PTH levels are highest around 2 AM and lowest around 2 PM.
• Collect a venous blood sample in a red-top tube. Note that some laboratories require blood in an iced plastic syringe.
• Obtain a serum calcium level determination at the same time if ordered. The serum PTH and serum calcium levels are important for a differential diagnosis.
After
• Indicate on the laboratory slip the time the blood was drawn, because a diurnal rhythm affects test results.
• Apply pressure or a pressure dressing to the venipuncture site.
• Check the venipuncture site for bleeding.
• Most PTH specimens are sent to a central laboratory. The specimen should be kept cold and transported on dry ice.
Test Results and Clinical Significance
Increased Levels
Hyperparathyroidism secondary to adenoma or carcinoma of the parathyroid gland: PTH is autonomously produced by the parathyroid gland. PTH levels are elevated.
Non–PTH-producing tumors (paraneoplastic syndrome) commonly noted with lung, kidney, or breast carcinoma: These tumors produce a “PTH-related protein” that acts like PTH and increases serum calcium. Because it is structurally similar to PTH, it is measured with PTH testing and gives a falsely high PTH result.
Congenital renal defect: These patients have a congenital kidney nonresponsiveness to “normal” quantities of PTH. As a result, calcium decreases despite normal quantities of PTH. The parathyroid is called on to produce even greater quantities of PTH. This is also called pseudohyperparathyroidism.
Hypocalcemia: PTH elevation is the result of a physiologic compensation for low serum calcium.
Chronic renal failure: These patients cannot excrete phosphates. As a result, serum calcium levels diminish. PTH elevation is the result of a physiologic compensation for low serum calcium. This is secondary hyperparathyroidism. Occasionally a patient with CRF develops parathyroid hyperplasia that leads to PTH levels in excess of the amount required for physiologic homeostasis. This is called tertiary hyperparathyroidism.
Malabsorption syndrome: These patients do not absorb calcium or fat-soluble vitamins such as vitamin D. Serum calcium falls. PTH elevation is the result of a physiologic compensation for low serum calcium.
Vitamin D deficiency and rickets: Vitamin D is integral to the absorption of calcium from the gut. With inadequate levels, serum calcium decreases. PTH elevation is the result of a physiologic compensation for low serum calcium.
Decreased Levels
Hypoparathyroidism caused by surgical ablation or immunoablation: PTH is not produced at levels necessary to maintain normal calcium levels.
Hypercalcemia: Reduced PTH is a normal physiologic response to high serum calcium.
Metastatic bone tumor: Tumor in the bone can mobilize large quantities of calcium. Reduced PTH is a normal physiologic response to high serum calcium.
Hypercalcemia of malignancy (most often with lung, breast, or lymphoma cancer): For unknown reasons, serum calcium levels become very high in these cancer patients. They do not have bone metastasis or PTH-related proteins. Reduced PTH is a normal physiologic response to high serum calcium.
Sarcoidosis: These patients can develop elevated serum calcium levels. Reduced PTH is a normal physiologic response to high serum calcium.
Vitamin D intoxication: These patients have high serum calcium levels as a result of vitamin D stimulating maximal calcium intestinal absorption. Reduced PTH is a normal physiologic response to high serum calcium.
Milk-alkali syndrome: These infants are given cooked whole milk that is very high in calcium. They develop high serum calcium levels. Reduced PTH is a normal physiologic response to high serum calcium.
DiGeorge syndrome: These immunodeficient children also have hypocalcemia that may be due to hypoparathyroidism.