Prostate Specific Antigen (PSA)
Normal Findings
Indications
This test is used as a screening method for early detection of prostatic cancer. When the PSA test is combined with a rectal examination, nearly 90% of clinically significant cancers can be detected. This test is also used to monitor the disease after treatment.
Test Explanation
PSA is a glycoprotein found in high concentrations in the prostatic lumen. Significant barriers such as prostate glandular tissue and vascular structure are interposed between the prostatic lumen and the bloodstream. These protective barriers can be broached when disease such as cancer, infection, and benign hypertrophy exists. PSA can be detected in all males; however, levels are greatly increased in patients with prostatic cancer.
Elevated PSA levels are associated with prostate cancer. Levels greater than 4 ng/mL have been found in more than 80% of men with prostate cancer. The higher the levels, the greater the tumor burden. The PSA assay is also a sensitive test for monitoring response to therapy. Successful surgery, radiation, or hormone therapy is associated with a marked reduction in the PSA blood level. Significant elevation in PSA subsequently indicates the recurrence of prostatic cancer. PSA is more sensitive and specific than other prostatic tumor markers, such as prostatic acid phosphatase (PAP). Also, PSA is more accurate than PAP in monitoring response to therapy and recurrence of tumor after therapy.
There is considerable controversy regarding the use of PSA screening among asymptomatic men. The US Preventative Services Task Force (USPSTF) and other professional societies have indicated that mortality from prostate cancer is not significantly reduced by annual PSA screening. Furthermore most feel that “PSA screening identified” prostate cancer is not an aggressive cancer and is not associated with a significant increase in mortality. Approximately 80% of PSA screening testing is falsely positive. A positive screening test often triggers a biopsy and even potential life-threatening surgery with very little benefit. However, PSA screening in high-risk men such as those of African-American descent, genetic predisposition (e.g., BRCA genetic mutation), or strong family history should be offered annual PSA testing and digital rectal examinations.
It is important to be aware that some patients with early prostate cancer will not have elevated levels of PSA. It is equally important to recognize that PSA levels above 4 are not always associated with cancer. The PSA is limited by a lack of specificity within the “diagnostic gray zone” of 4 to 10 ng/mL. PSA levels also may be minimally elevated in patients with benign prostatic hypertrophy (BPH) and prostatitis. In an effort to increase the accuracy of PSA testing, other measures of PSA (Box 2-17) have been proposed.
• PSA velocity: PSA velocity is the change in PSA levels over time. A sharp rise in the PSA level raises the suspicion of cancer and may indicate a fast-growing cancer. Men who had a PSA velocity above 0.35 ng/mL per year had a higher relative risk for dying from prostate cancer than men who had a PSA velocity less than 0.35 ng/mL per year.
• Age-adjusted PSA (Table 2-40): Age is an important factor in increasing PSA levels. Men younger than age 50 should have a PSA level below 2.4 ng/mL, whereas a PSA level up to 6.5 ng/mL would be considered normal for men in their 70s.

• PSA density: PSA density considers the relationship of the PSA level to the size of the prostate. The use of PSA density to interpret PSA results is controversial because cancer might be overlooked in a man with an enlarged prostate. PSA density is an adjustment that divides the PSA measurement by the gland volume. Several formulas have been created to partially correct for gland volume. One such volume adjusted formula is:

• Free versus bound PSA: PSA circulates in the blood in two forms: free or bound to a protein molecule. With benign prostate conditions (such as BPH), there is more free PSA, while cancer produces more of the bound form. If a man's attached PSA is high but his free PSA is not, the presence of cancer is more likely. When the %FPSA is less than 25%, there is a high likelihood of cancer (Table 2-41).

• Alteration of PSA cutoff level: Some researchers have suggested lowering the cutoff levels that determine if a PSA measurement is normal or elevated. For example, a number of studies have used cutoff levels of 2.5 or 3.0 ng/mL (rather than 4.0 ng/mL).
• Prostate-specific proteins: Patterns of prostate proteins are being studied to determine if a biopsy is necessary when a person has a slightly elevated PSA level or an abnormal DRE. Prostatic specific membrane antigen may, with further study, represent an excellent marker for prostate cancer. It is more frequently present than PSA in more advanced cancer. Another protein of interest is Early Prostate Cancer Antigen (EPCA). Unlike the PSA, this protein is not found in normal prostate cells. Instead, EPCA occurs in relatively large amounts only in prostate cancer cells. Early testing suggests that EPCA may be more accurate than PSA in identifying prostate cancer. Furthermore, EPCA levels are significantly higher in patients whose cancers spread outside the prostate compared with those with disease confined to the gland. EPCA-1 is a tissue-based test and EPCA-2 is a blood-based test. Patients with an EPCA-2 cutoff level of 30 ng/mL or higher are considered to be at risk for prostate cancer.
• Prostate cancer specific biomarkers: These biomarkers are made up of RNA that is present in prostate cancer cells at very high levels because of overexpression of particular genes. These biomarkers can be detected in the urine of patients with prostate cancer after a short period of professional prostate massage. The most commonly tested marker is the prostate cancer gene 3 (PCA3). Other genetic markers tested include GOLPH2, SPINK1, and TMPRSS2-ERG. These biomarkers are not elevated in noncancerous prostate disease. Furthermore these biomarkers are not influenced by patient age or prostate volume.
PSA is used in the staging of men with known prostate cancer. Men with PSA levels below 10 ng/mL are most likely to have localized disease and respond well to local therapy (radical prostatectomy or radiation therapy). Routine metastatic staging tests are generally not required for men with clinically localized prostate cancer when their PSA is less than 20 ng/mL.
PSA is used to follow up men after treatment for prostate cancer. Periodic PSA testing should follow any form of treatment for prostate cancer, since PSA levels can indicate need for further treatment. Following curative radical prostatectomy or radiation therapy, PSA levels should probably be 0 to 0.5 ng/mL. The pattern of PSA rise after local therapy for prostate cancer can help distinguish between local recurrence and distant spread. Patients with elevated PSA levels more than 24 months after local treatment and with a PSA doubling time after 12 months are likely to have recurrence.
PSA can be measured by electrochemiluminescent immunoassay, immunohistochemistry, or radioimmunoassay. Newer, comparably accurate, chemical tests are being used to improve the worldwide use of PSA screening testing.
Interfering Factors
• Rectal examinations are well known to falsely elevate PAP levels, and they may also minimally elevate the PSA. To avoid this problem, the PSA should be drawn before rectal examination of the prostate or several hours afterward.
• Prostatic manipulation by biopsy or transurethral resection of the prostate (TURP) will significantly elevate the PSA levels. The blood test should be done before surgery or 6 weeks after manipulation.
• Ejaculation within 24 hours of blood testing will be associated with elevated PSA levels.
• Recent urinary tract infection or prostatitis can cause elevations of PSA as much as five times baseline for as long as 6 weeks.
 Finasteride (Propecia, Proscar) and diethylstilbesterol (DES) may cause decreased levels of PSA.
Finasteride (Propecia, Proscar) and diethylstilbesterol (DES) may cause decreased levels of PSA.
Procedure and Patient Care
Related Test
Prostatic Acid Phosphatase (PAP) (p. 25). This is another tumor marker for prostate cancer. It is less specific and less sensitive than the PSA test, and its use is diminishing.
Protein (Protein Electrophoresis, Immunofixation Electrophoresis [IFE], Serum Protein Electrophoresis [SPEP], Albumin, Globulin, Total Protein)
Normal Findings
Indications
The measurement of proteins is a part of most routine screening tests. Protein electrophoresis, however, is used to identify protein abnormalities caused by a wide spectrum of diseases, including infections, inflammation, and hematologic malignancy.
Test Explanation
Proteins are constituents of muscle, enzymes, hormones, transport vehicles, hemoglobin, and several other key functional and structural entities within the body. They are the most significant components contributing to the osmotic pressure within the vascular space. This osmotic pressure keeps fluid within the vascular space, minimizing extravasation of fluid.
Albumin and globulin constitute most of the protein within the body and are measured together as the total protein. Albumin is a protein that is formed within the liver. It makes up approximately 60% of the total protein. The major effect of albumin within the blood is to maintain colloidal osmotic pressure. Furthermore, albumin transports important blood constituents such as drugs, hormones, and enzymes. Albumin is synthesized within the liver and is therefore a measure of hepatic function. When disease affects the liver cell, the hepatocyte loses its ability to synthesize albumin. The serum albumin level is greatly decreased. Because the half-life of albumin is 12 to 18 days, however, severe impairment of hepatic albumin synthesis may not be recognized until after that period.
Globulins represent all non-albumin proteins. Their role in maintaining osmotic pressure is far less than that of albumin. Alphal globulins are mostly alpha1 antitrypsin. Some transporting proteins, such as thyroid and cortisol-binding globulin, also contribute to this electrophoretic zone. Alpha2 globulins include serum haptoglobins (which bind hemoglobin during hemolysis), ceruloplasmin (which is a carrier for copper), prothrombin, and cholinesterase (which is an enzyme used in the catabolism of acetylcholine). Beta1 globulins include lipoproteins, transferrin, plasminogen, and complement proteins; beta2 globulins include fibrinogen. Gamma globulins are the immunoglobulins (antibodies) (p. 312). To a lesser degree, globulins also act as transport vehicles.
Serum albumin and some globulins are measures of nutrition. Malnourished patients, especially after surgery, have a greatly decreased level of serum proteins. Burn patients and those who have protein-losing enteropathies and uropathies, have low levels of protein despite normal synthesis. Pregnancy, especially in the third trimester, is usually associated with reduced total proteins.
In some diseases, albumin is selectively diminished, and globulins are normal or increased to maintain a normal total protein level. For example, in collagen vascular diseases (e.g., lupus erythematosus), capillary permeability is increased. Albumin, a molecule that is generally smaller than most globulins, is selectively lost into the extravascular space. Another group of diseases similarly associated with low albumin, high globulin, and normal total protein levels is chronic liver diseases. In these diseases the liver cannot produce albumin, but globulin is adequately made in the reticuloendothelial system. In both of these types of diseases the albumin level is low, but the total protein level is normal because of increased globulin levels. These changes, however, can be detected if one measures the albumin/globulin ratio. Normally this ratio exceeds 1.0. The diseases just described that selectively affect albumin levels are associated with lesser ratios. Increased total protein levels, particularly the globulin fraction, occur with multiple myeloma and other gammopathies. It is important to note that proteins can be factitiously elevated in dehydrated patients. This is particularly well documented by measurement of the albumin level. Albumin, globulin, and other proteins can be quantitated individually. See specific protein tests.
Serum protein electrophoresis (SPEP) can separate the various components of blood protein into bands or zones according to their electrical charge. Several well-established electrophoretic patterns have been identified and can be associated with specific diseases (Table 2-42). If a spike is detected, immunofixation techniques can be added to the electrophoretic strip. In general, polyclonal spikes are associated with infectious or inflammatory diseases in which monoclonal specific spikes are often neoplastic. Immunofixation is used to indicate deficiencies or excesses as seen with macroglobulinemia, monoclonal gammopathy of undetermined significance (MGUS), and multiple myeloma. Immunofixation is also able to determine whether a monoclonal spike is caused by light-chain or other protein abnormalities.
TABLE 2-42
Protein Electrophoresis Patterns in Specific Diseases
| Pattern | Electrophoresis | Disease |
| Acute reaction | ↓ Albumin ↑ Alpha2 globulin |
Acute infections, tissue necrosis, burns, surgery, stress, myocardial infarction |
| Chronic inflammation | sl. ↓ Albumin sl. ↑ Gamma globulin N Alpha2 globulin |
Chronic infection, granulomatous diseases, cirrhosis, rheumatoid-collagen diseases |
| Nephrotic syndrome | ↓↓ Albumin ↑↑ Alpha2 globulin N ↑ Beta globulin |
Nephrotic syndrome |
| Far-advanced cirrhosis | ↓ Albumin ↑ Gamma globulin Incorporation of beta and gamma peaks |
Far-advanced cirrhosis |
| Polyclonal gamma globulin elevation | ↑↑ Gamma globulin with a broad peak | Cirrhosis, chronic infection, sarcoidosis, tuberculosis, endocarditis, rheumatoid-collagen diseases |
| Hypogammaglobulinemia | ↓ Gamma globulin with normal other globulin levels | Light-chain multiple myeloma |
| Monoclonal gammopathy | Thin spikes in the beta (IgA, IgM) and gamma globulins | Myeloma, Waldenström macroglobulinemia, gammopathies |
↓, Decreased; ↑, increased; sl. ↓, slightly decreased; sl. ↑, slightly increased; N, normal; ↓↓, greatly decreased; ↑↑, greatly increased.
With immunofixation, a monospecific antibody is placed in contact with the gel after the proteins have been separated by electrophoresis. The resulting protein-antibody complexes are subsequently specifically stained for visualization after being precipitated out. The pathologist can then identify and classify specific immunoglobulin spikes. Specific monoclonal protein studies can be performed on the urine or blood. Monoclonal immunoglobulin heavy chain (gamma, alpha, mu, delta, or epsilon) and/or light chains (kappa or lambda) can be identified. With sensitive nephelometric assay specific light chain disease can be identified (Figures 2-21 through 2-25).

Figure 2-21 Normal automated serum protein electrophoresis for patients 1 through 7. Note dense albumin electrophoresis on top followed by globulins toward the bottom.
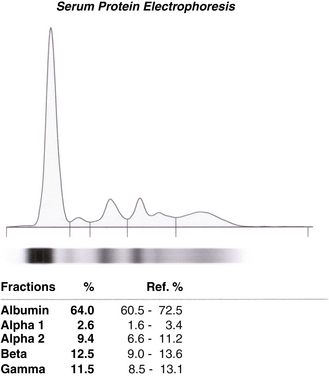
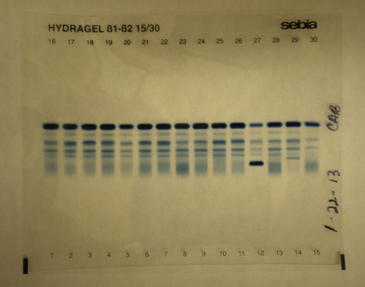
Figure 2-23 Abnormal automated serum protein electrophoresis for patients 1 through 10. Note dense migration of the paraprotein for patient 4.
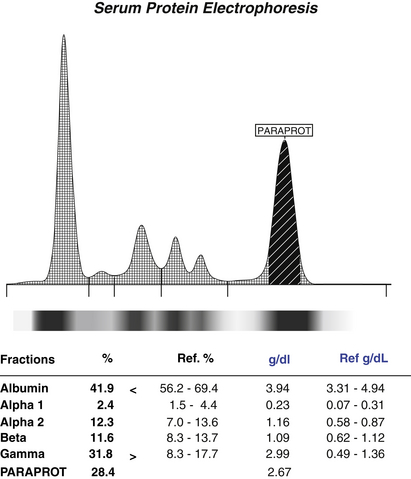
Figure 2-24 Normal automated serum protein electrophoresis in graphic form for patient 4. An abnormal globulin paraprotein is noted.

Figure 2-25 Abnormal immunofixation immunoglobulin electrophoresis for patient 4. ELP equals protein electrophoresis pattern. Note the dense migration pattern in the lower portion of the ELP column. G equals IgG antibody; A equals IgA antibody; M equals IgM antibody; K equals kappa chains; L equals lambda chains. This patient has an IgA and lambda chain gammopathy.
This test is also used to follow the course of the disease or treatment in patients with known monoclonal immunoglobulinopathies. For example, with successful treatment for neoplastic gammopathies, IFE, upon repetition, can demonstrate reduction in the specific immunoglobulin. Finally, this test is helpful in defining more clearly the immune status of a patient whose immune status may be compromised.
Protein electrophoresis is also used to evaluate the major protein fractions found in urine. Normally only small amounts of albumins are seen. Urinary protein electrophoresis is useful in classifying the type of renal damage, if present. Immunofixation is useful in characterizing M-components observed in the protein electrophoresis and in identifying light-chain disease. These electrophoresis techniques can be provided to the CSF or any body fluid.
Interfering Factors
• Prolonged application of tourniquet can increase both fractions of total proteins.
• Sampling of peripheral venous blood proximal to an IV administration site can result in an inaccurately low protein level. Likewise, massive IV infusion of crystalloid fluid can result in acute hypoproteinemia.
Drugs that can cause increased protein levels include anabolic steroids, androgens, corticosteroids, dextran, growth hormone, insulin, phenazopyridine, and progesterone.
Drugs that can cause decreased protein levels include ammonium ions, estrogens, hepatotoxic drugs, and oral contraceptives.
Procedure and Patient Care
Test Results and Clinical Significance
 Decreased Albumin Levels
Decreased Albumin Levels
Malnutrition: Lack of amino acids available for building proteins contributes to this observation. Probably the liver dysfunction (albumin synthesis) associated with malnutrition also contributes to the low albumin levels.
Pregnancy: Albumin levels progressively decrease until delivery.
Liver disease (e.g., hepatitis, extensive metastatic tumor, cirrhosis, hepatocellular necrosis): The liver is the site of synthesis of albumin. If production of albumin is inadequate, levels can be expected to fall.
Protein-losing enteropathies (e.g., malabsorption syndromes such as Crohn disease, sprue, Whipple disease): Large volumes of protein are lost from the intestines because absorption is inadequate. Albumin levels will fall.
Protein-losing nephropathies (e.g., nephrotic syndrome, nephrosis): Large volumes of albumin can be lost through the kidneys. This loss may be selective for albumin (lipoid nephrosis) or drain out all components of proteins (glomerulonephritis).
Third-space losses (e.g., ascites, third-degree burns): Large amounts of albumin can be lost in the serum that weeps from chronic open burns. Albumin readily accumulates in the peritoneum of patients with ascites.
Overhydration: As the blood volume increases, albumin concentration measurements decrease mathematically.
Increased capillary permeability (e.g., collagen-vascular diseases such as lupus erythematosus): Albumin can seep out of the microvascular spaces in the tissues and cause edema or in the kidneys and cause proteinuria. The serum albumin decreases.
Inflammatory disease: Diseases associated with inflammation, necrosis, infarction, or burns cause an increase in acute-phase reactant proteins. These are mostly globulins. Therefore the globulin component of proteins increases and albumin decreases.
Familial idiopathic dysproteinemia: This is a genetic disease in which albumin is significantly reduced (and globulins are increased).
Decreased Alpha1 Globulin Levels
Juvenile pulmonary emphysema: These patients have a genetic decrease or absence of alpha1 antitrypsin, which is important to normal pulmonary function.
Decreased Alpha2 Globulin Levels
Hemolysis: Haptoglobin is an alpha2 globulin and is decreased when hemolysis occurs.
Wilson disease: Ceruloplasmin is an alpha2 globulin. It is decreased in Wilson disease.
Severe liver dysfunction: Haptoglobulin is an alpha2 globulin that is made in the liver. It is decreased when liver function is inadequate.
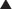 Increased Beta Globulin Levels
Increased Beta Globulin Levels
Hypercholesterolemia (which can occur by itself or in association with biliary cirrhosis, hypothyroidism, or nephrosis): Beta lipoprotein is a beta globulin and is increased in hypercholesterolemia.
Iron-deficiency anemia: Transferrin is a beta globulin and is increased in this form of anemia.
Estrogen therapy: Estrogen causes increased production of these proteins.
Increased Gamma Globulin Levels
Waldenström macroglobulinemia:
These cancers are characterized by production of gamma globulin from neoplastic plasma cells or lymphocytes. The total gamma globulin zone may not be increased but a monoclonal spike in one portion is often seen.
Chronic inflammatory disease (e.g., rheumatoid arthritis, systemic lupus erythematosus [SLE]): These diseases are associated with autoantibodies, and patients will have a gamma globulin spike.
Malignancy (e.g., Hodgkin's disease, lymphoma, leukemia): These diseases may be associated with elevated gamma globulins.
Hyperimmunization: A small spike can occur in the IgA portion of the gamma band.
Cirrhosis: Most patients have gamma and some have beta globulin spikes associated with this disease. The pathophysiology is not well known.
Acute and chronic infection: Infection is associated with an antibody response and therefore an increase in immunoglobulins (gamma globulins).
Decreased Gamma Globulin Levels
Genetic immune disorders: A host of immune deficiencies are associated with reduced or absent immunoglobulins.
Secondary immune deficiency: Several conditions (e.g., steroid use, nephrotic syndrome, severe gram-negative infection, lymphoma, leukemia) are associated with deficient levels of immunoglobulins.
Increased Urine Monoclonal Immunoglobulins
Waldenström's macroglobulinemia:
These diseases are highlighted by rapid cellular duplication of mononuclear antibody producing cells.
See also Table 2-42.
Related Tests
Immunoglobulin Quantification (p. 312). This is a measurement of each various immunoglobulin and a determination of its clonality.
Protein C, Protein S
Test Explanation
The plasma coagulation system is tightly regulated between thrombosis and fibrinolysis. This precise regulation is important. The protein C–protein S system is an important inhibitor of coagulation. Protein C inhibits the activation of factors VIII and V (see Figure 2-12, p. 167). This inhibitory function of protein C is enhanced by protein S. Congenital deficiencies of these vitamin K–dependent proteins may cause spontaneous intravascular thrombosis. Furthermore dysfunctional forms of the proteins result in a hypercoagulable state. In addition nearly 50% of hypercoagulable states are caused by the presence of a factor V (factor V-Leiden, p. 231) that is resistant to protein C inhibition. Acquired deficiencies are less commonly symptomatic.
When protein C is tested, protein S activity also should be tested because the decreased activity of protein C may be the result of decreased protein S. When decreased protein C activity is noted, protein C resistance (the presence of factor V-Leiden) should be tested.
These proteins are vitamin K dependent and are decreased in patients who are taking Coumadin, as well as those with liver diseases or severe malnutrition. Of the total plasma protein S, approximately 60% circulates bound to C4bBP compliment protein, whereas the remaining 40% circulates as “free” protein S. Only free protein S has an anticoagulant function. Because compliment regulatory proteins are acute phase reactants, autoimmune diseases and other inflammatory diseases are associated with increased binding of protein S, causing an acquired protein S deficiency. Affected patients may experience hypercoagulable events.
Measurement of plasma free protein S antigen is performed as the initial testing for protein S deficiency. When the free protein S antigen level is below the age- and sex-adjusted normal range, quantification for total plasma protein S antigen is indicated.
Interfering Factors
• Decreased protein C may occur in the postoperative states.
• Pregnancy or the use of exogenous sex hormones is associated with decreases in proteins C and S. These low levels of protein S in pregnancy do not cause thrombosis by themselves.
• The concentration of citrate in the collection tube varies and can affect activity results.
• Active clotting states, such as DVT, can lower levels of protein S and C.
 Drugs that can decrease levels include vitamin K inhibitors such as coumadin.
Drugs that can decrease levels include vitamin K inhibitors such as coumadin.
Procedure and Patient Care
During
• Collect a venous blood sample in a blue-top tube. If more than one blood test is to be obtained, draw the blood for proteins C or S second to avoid contamination with tissue thromboplastin that may occur in the first tube. If only blood for protein C or S is being drawn, draw a red top first (and throw it away) and then draw the blood for this study in a blue top tube (two-tube method of blood draw).
Test Results and Clinical Significance
Decreased Levels
Inherited deficiency of protein C or protein S: Protein S or C defect that may not be recognized until adulthood.
Disseminated intravascular coagulation (DIC),
Arterial or venous thrombosis:
Vitamin K deficiency: Protein C and S are dependent on vitamin K for their synthesis. If vitamin K is not available because of malnutrition, biliary disease, or malabsorption, these proteins will not be produced in adequate levels. Because several coagulation factors are also vitamin K-dependent, a hypercoagulable event may not occur.
Sickle cell disease: This condition alone does not produce a thrombophilic state.
Coumadin-induced skin necrosis: This occurs in the feet, buttocks, thighs, breasts, upper extremities, and genitalia. The lesions usually begin as maculopapular lesions several days after initiation of warfarin and progress into bullous, hemorrhagic, necrotic lesions. Patients with protein C deficiency are at high risk for warfarin-induced skin necrosis during initiation of therapy with warfarin. Approximately one third of patients with warfarin-induced skin necrosis have protein C deficiency.
Related Test
Disseminated Intravascular Coagulation (DIC) Screening (p. 210). This group of tests is indicated for patients with coagulopathies, such as DIC.
Prothrombin Time (PT, Pro-Time, International Normalized Ratio [INR])
Indications
The PT is used to evaluate the adequacy of the extrinsic system and common pathway in the clotting mechanism.
Test Explanation
The hemostasis and coagulation system is a homeostatic balance between factors encouraging clotting and the factors encouraging clot dissolution. The first reaction of the body to active bleeding is blood vessel constriction. In small vessel injury, this may be enough to stop bleeding. In large vessel injury, hemostasis is required to form a clot that will durably plug the hole until healing can occur. The primary phase of the hemostatic mechanism involves platelet aggregation to blood vessel (see Figure 2-12 on p. 167). Next, secondary hemostasis occurs. The first phase of reactions is called the intrinsic system. Factor XII and other proteins form a complex on the subendothelial collagen in the injured blood vessel. Through a series of reactions, activated factor XI (XIa) is formed and activates factor IX (IXa). In a complex formed by factors VIII, IX, and X, activated X (Xa) is formed.
At the same time, the extrinsic system is activated and a complex is formed between tissue thromboplastin (factor III) and factor VII (which is exposed after cellular injury). Activated factor VII (VIIa) results. Factor VIIa can directly activate factor X. Alternatively, VIIa can activate IX and X together.
In the third phase, factor X is activated by the proteases formed by the two prior reactions and by activated factor IX. This reaction is a common pathway that provides the link between the intrinsic and the extrinsic systems. In the fourth and final phase, prothrombin is converted into thrombin by activated factor X in the presence of factor V, phospholipid, and calcium.
Thrombin not only converts fibrinogen to fibrin in “clot stabilization” but also stimulates platelet aggregation and activates factors V, VIII, and XIII. Once fibrin is formed, it is then polymerized into a stable gel. Factor XIII cross-links the fibrin polymers to form a stable clot.
Almost immediately three major activators of the fibrinolytic system act on plasminogen, which was previously absorbed into the clot, to form plasmin. Plasmin degenerates the fibrin polymer into fragments, which are cleared by macrophages.
The PT measures the clotting ability of factors I (fibrinogen), II (prothrombin), V, VII, and X (i.e., the extrinsic system and common pathway). When these clotting factors exist in deficient quantities, the PT is prolonged. Many diseases and drugs are associated with decreased levels of these factors. These include the following:
1. Hepatocellular liver disease (e.g., cirrhosis, hepatitis, and neoplastic invasive processes). Factors I, II, V, VII, IX, and X are produced in the liver. With severe hepatocellular dysfunction, synthesis of these factors will not occur, and serum concentration of these factors will be decreased.
2. Obstructive biliary disease (e.g., bile duct obstruction secondary to tumor or gallstones or intrahepatic cholestasis secondary to sepsis or drugs). As a result of the biliary obstruction, the bile necessary for fat absorption fails to enter the gut, and fat malabsorption results. Vitamins A, D, E, and K are fat soluble and also are not absorbed. Because the synthesis of factors II, VII, IX, and X depends on vitamin K, these factors will not be adequately produced, and serum concentrations will fall. Hepatocellular liver disease can be differentiated from obstructive biliary disease by determination of the patient's response to parenteral vitamin K administration. If the PT returns to normal after 1 to 3 days of vitamin K administration (10 mg intramuscularly twice a day), one can safely assume that the patient has obstructive biliary disease that is causing vitamin K malabsorption. If, on the other hand, the PT does not return to normal with the vitamin K injections, one can assume that severe hepatocellular disease exists and that the liver cells are incapable of synthesizing the clotting factors no matter how much vitamin K is available.
3. Oral anticoagulant administration. The coumarin derivatives dicumarol and warfarin (Coumadin, Panwarfin) are used to prevent coagulation in patients with thromboembolic disease (e.g., pulmonary embolism, thrombophlebitis, arterial embolism). These drugs interfere with the production of vitamin K–dependent clotting factors, which results in a prolongation of PT, as already described. The adequacy of coumarin therapy can be monitored by following the patient's PT. For anticoagulation, the INR typically should be between 2.0 and 3.0 for patients with atrial fibrillation, and between 3.0 and 4.0 for patients with mechanical heart valves. However, the ideal INR must be individualized for each patient (Table 2-43).

PT test results used to be given in seconds, along with a control value. The control value usually varied somewhat from day to day because the reagents used varied. The patient's PT value was supposed to be approximately equal to the control value. Some laboratories used to report PT values as percentages of normal activity, because the patient's results were compared with a curve representing normal clotting time. A normal PT result was 85% to 100%.
To have uniform PT results for physicians in different parts of the country and the world, the World Health Organization has recommended that PT results include the use of the international normalized ratio (INR) value. The reported INR results are independent of the reagents or methods used. Many hospitals are now reporting PT times in both absolute and INR numbers. Factors such as weight, body mass index, age, diet, and concurrent medications are known to affect warfarin dose requirements during anticoagulation therapy.
Warfarin interferes with the regeneration of reduced vitamin K from oxidized vitamin K in the VKOR (vitamin K oxidoreductase) complex. A recently identified gene for the major subunit of VKOR, called VKORC1, has been identified and may explain up to 44% of the variance in warfarin dose requirements. Furthermore, warfarin is metabolized in part by the cytochrome P-450 enzyme CYP2C9. The CYP2C9∗2 and CYP2C9∗3 genetic mutations have been shown to decrease the enzyme activity of these metabolizing enzymes, which has led to warfarin sensitivity and, in serious cases, bleeding complications. A warfarin pharmacogenomic test panel is available that can identify any mutations in the VKORC1-1639, CYP2C9∗2, or CYP2C9∗3 genes. The warfarin pharmacogenomic test can be used as part of an algorithm to determine the best initial warfarin dose and does not replace the need for routine PT testing for the calculation of the INR.
Point-of-care home testing is now available for patients who require long-term anticoagulation with warfarin. This is useful for patients with prosthetic cardiac valves, chronic atrial fibrillation, or recurrent venous thromboembolism, and is especially helpful for patients who do not live close to a testing facility. Like glucose monitoring, a finger stick is performed. A drop of blood is placed on the testing strip and inserted into the handheld testing device. The PT and INR are provided in a few minutes. The treating physician is notified by phone and any therapeutic changes can be instigated the same day.
Coumarin derivatives are slow acting, but their action may persist for 7 to 14 days after discontinuation of the drug. The action of a coumarin drug can be reversed in 12 to 24 hours by slow parenteral administration of vitamin K (phytonadione). The administration of plasma will even more rapidly reverse the coumarin effect. The action of coumarin drugs can be enhanced by drugs such as aspirin, quinidine, sulfa, and indomethacin. Barbiturates, chloral hydrate, and oral contraceptives cause increased coumarin drug binding and therefore may decrease the effects of coumarin drugs.
Interfering Factors
• Alcohol intake can prolong PT times. Alcohol diminishes liver function. Many factors are made in the liver. Lesser quantities of coagulation factors result in prolonged PT times.
• A diet high in fat or leafy vegetables may shorten PT times. Absorption of vitamin K is enhanced. Vitamin K–dependent factors are made at increased levels, thereby shortening PT times.
• Diarrhea or malabsorption syndromes can prolong PT times. Vitamin K is malabsorbed, and as a result, factors II, VII, IX, and X are not made.
Drugs that may cause increased levels include allopurinol, aminosalicylic acid, barbiturates, beta-lactam antibiotics, chloral hydrate, cephalothins, cholestyramine, cimetidine, clofibrate, colestipol, ethyl alcohol, glucagon, heparin, methyldopa, neomycin, oral anticoagulants, propylthiouracil, quinidine, quinine, salicylates, and sulfonamides.
Drugs that may cause decreased levels include anabolic steroids, barbiturates, chloral hydrate, digitalis, diphenhydramine, estrogens, griseofulvin, oral contraceptives, and vitamin K.
Procedure and Patient Care
Before
 Explain the procedure to the patient.
Explain the procedure to the patient.
Tell the patient that no fasting is required.
• If the patient is receiving warfarin, obtain the blood specimen before the patient is given the daily dose of warfarin. The daily dose may be increased, decreased, or kept the same depending on the PT test results for that day.
After
• Apply pressure or a pressure dressing to the venipuncture site.
• Assess the venipuncture site for bleeding. Remember, hemostasis will be delayed if the patient is taking warfarin or if the patient has any coagulopathies.
• If the PT is greatly prolonged, evaluate the patient for bleeding tendencies (i.e., check for blood in the urine and all excretions and assess the patient for bruises, petechiae, and low-back pain). Back pain may be a symptom of retroperitoneal bleeding.
• If severe bleeding occurs, the anticoagulant effect of warfarin can be reversed by the slow parenteral administration of vitamin K (phytonadione). If coagulation must be returned to near normal more quickly, plasma can be given.
Because of drug interactions, instruct the patient not to take any medication unless specifically ordered by the physician.
 Home Care Responsibilities
Home Care Responsibilities
• Coumadin levels will be regulated by PT and INR values.
• Inform patients to evaluate themselves for bleeding tendencies. Patients should assess themselves for bruises, petechiae, low-back pain, and bleeding gums. Blood may be detected in the urine and stool.
• Because of many drug interactions, instruct patients on coumadin therapy not to take any other medications unless approved by their physician.
Test Results and Clinical Significance
Increased Levels (Prolonged PT)
Liver disease (e.g., cirrhosis, hepatitis): Coagulation factors are made in the liver. With liver disease, synthesis is inadequate and the PT is increased.
Hereditary factor deficiency: A genetic defect causes a decrease in a coagulation factor. The PT is increased. Factors II, V, VII, or X could be similarly affected.
Vitamin K deficiency: Vitamin K–dependent factors (II, VII, IX, X) are not made. The PT is increased.
Bile duct obstruction: Fat-soluble vitamins, including vitamin K, are not absorbed. Vitamin K–dependent factors (II, VII, IX, X) are not made. The PT is increased.
Coumarin ingestion: Synthesis of the vitamin K–dependent coagulation factors is inhibited. The PT is increased.
Disseminated intravascular coagulation (DIC): Coagulation factors are consumed in the intravascular coagulation process. The PT is increased.
Massive blood transfusion: Coagulation is inhibited by the anticoagulant in the banked blood. Furthermore, with massive bleeding the factors are diluted out by the “factor-poor” banked blood.
Rabies-Neutralizing Antibody
Indications
This test is performed after vaccination to document seroprotection in animal care workers. It is also used to determine exposure to rabies and in the diagnosis of rabies.
Test Explanation
Identification and documentation of the presence of rabies-neutralizing antibody is important for veterinary health care workers and others who are at risk or may have been exposed to the rabies virus. This test is performed on persons who are at great risk for animal bites (veterinarians and their staff, zoo workers, those who work with animals in laboratories) and on those who have received the human diploid cell rabies vaccine (HDCV). A rabies titer of greater than 1:16 is considered protective.
Rabies antibody is also used in diagnosing rabies in patients suspected of being exposed to the virus. A fourfold rise in antibody titer over several weeks in a person not previously exposed to the HDCV indicates rabies exposure. If the patient has received HDCV and has been bitten by an animal suspected of having rabies infection, a very high antibody titer may support the diagnosis. The presence of antibody in the cerebrospinal fluid (CSF) is also supportive of the diagnosis, because usually there are not antibodies in the CSF after the HDCV vaccine, but there are antibodies after a bite from a rabies-infected animal. In patients who may have been exposed to rabies, the human rabies immunoglobulin (HRIG) is given after the antibody titers have been obtained. Half of the HRIG is given into the area of the bite, and half is administered as an intramuscular (IM) injection into the gluteal region. At the same time the first of the HDCV shots are administered to begin vaccination. Four subsequent IM injections are administered over the next 28 days. One can expect to see increases in rabies antibody levels in about 10 days, but protective levels may not be present for several weeks. Postexposure protocols exist to determine the proper handling of the patient and animal, depending on the real risk for the animal's infection.
The rabies antibody is identified by the direct fluorescent antibody method. More recently immunofluorescence has been used.
Test Results and Clinical Significance
Exposure to rabies vaccine: This causes a relatively low titer of 1:16 or greater.
Recent bite exposure to rabies virus: This causes a progressive rise in titer to levels of 1:200 to 1:160,000.
Active rabies in patient or animal: Antibody titers are extremely high in patients who present with encephalitis and brain stem dysfunction. These patients rarely recover from the disease.
Red Blood Cell Count (RBC Count, Erythrocyte Count)
Indications
The RBC count is closely related to the hemoglobin (p. 281) and hematocrit (p. 277) levels and represents different ways of evaluating the number of RBCs in the peripheral blood. It is repeated serially in patients with ongoing bleeding or as a routine part of the complete blood cell count. It is an integral part of the evaluation of anemic patients.
Test Explanation
This test is a count of the number of circulating RBCs in 1 mm3 of peripheral venous blood. The RBC count is routinely performed as part of a complete blood cell count. Within each RBC are molecules of hemoglobin that permit the transport and exchange of oxygen to the tissues and carbon dioxide from the tissues. The RBC is produced by the erythroid elements in the bone marrow. Under the stimulation of erythropoietin, RBC production is increased. Normally RBCs survive in the peripheral blood for approximately 120 days. During that time the RBC is transported through the bloodstream. In the smallest of capillaries the RBC must fold and bend to conform to the size of these tiny vessels. Toward the end of the RBC's life, the cell membrane becomes less pliable; the aged RBC is then lysed and extracted from the circulation by the spleen. Abnormal RBCs have a shorter life span and are extracted earlier. Intravascular RBC trauma, such as that caused by artificial heart valves or peripheral vascular atherosclerotic plaques, also shortens the RBC's life. An enlarged spleen, such as that caused by portal hypertension or leukemia, may inappropriately destroy and remove normal RBCs from the circulation.
Normal RBC values vary according to gender and age. Women tend to have lower values than men, and RBC counts tend to decrease with age. When the value is decreased below the range of the expected normal value, the patient is said to be anemic. Low RBC values are caused by many factors, including:
1. Hemorrhage (as in GI bleeding or trauma)
2. Hemolysis (as in glucose-6-phosphate dehydrogenase deficiency, spherocytosis, or secondary splenomegaly)
3. Dietary deficiency (as of iron or vitamin B12)
4. Genetic aberrations (as in sickle cell anemia or thalassemia)
5. Drug ingestion (as of chloramphenicol, hydantoins, or quinidine)
6. Marrow failure (as in fibrosis, leukemia, or antineoplastic chemotherapy)
RBC counts greater than normal can be physiologically induced as a result of the body's requirements for greater oxygen-carrying capacity (e.g., at high altitudes). Diseases that produce chronic hypoxia (e.g., congenital heart disease) also provoke this physiologic increase in RBCs. Polycythemia vera is a neoplastic condition causing uncontrolled production of RBCs.
Like the hemoglobin and hematocrit values, the RBC count can be altered by many factors other than RBC production. For instance, in dehydrated patients the total blood volume is contracted. The RBCs will be more concentrated, and the RBC count will be falsely high. Likewise, in overhydrated patients the blood concentration is diluted and the RBC count per millimeter will be falsely low. In most hospitals and laboratories the RBC count is done by an automated counting machine with an error range of about 4% to 5%.
Interfering Factors
• Normal RBC decreases are seen during pregnancy as a result of normal body fluid increases that dilute the RBCs. Also, there is an element of nutritional deficiency that is often associated with pregnancy that may play a role in the anemia of pregnancy.
• Living in high altitudes causes increased RBC counts as a result of a physiologic response to the decreased oxygen available at these high altitudes.
• Hydration status: As stated above, dehydration factitiously increases the RBC count, and overhydration decreases the RBC count.
Drugs that may cause increased RBC levels include erythropoietin and gentamicin.
Drugs that may cause decreased RBC levels include those that decrease marrow production or cause hemolysis.
Test Results and Clinical Significance
Increased Levels
Erythrocytosis: The number of RBCs increases as a result of illnesses or as a physiologic response to external situations (e.g., high altitude).
Congenital heart disease: Cyanotic heart diseases cause chronically low Po2 levels. In response, the RBCs increase in number.
Severe chronic obstructive pulmonary disease (COPD): Chronic states of hypoxia cause stimulation of RBC production as a physiologic response to increase oxygen-carrying capacity.
Polycythemia vera: This is a result of the bone marrow inappropriately producing great numbers of RBCs.
Severe dehydration (e.g., severe diarrhea or burns): With depletion of extracellular fluid, the total blood volume decreases, but the number of RBCs stays the same. Because the blood is more concentrated, the number of RBCs per cubic millimeter is increased.
Decreased Levels
Anemia: This is a state associated with reduced RBC numbers. Many different types of diseases are associated with anemia.
Hemoglobinopathy: Patients with hemoglobin disorders or other blood dyscrasias may have a reduced RBC number and survival.
Cirrhosis: This is a chronic state of fluid overload. The RBCs are diluted, and the number of RBCs per cubic millimeter is reduced.
Hemolytic anemia (e.g., erythroblastosis fetalis, hemoglobinopathies, drug-induced hemolytic anemias, transfusion reactions, paroxysmal nocturnal hemoglobinuria): The RBC survival is diminished in hemolytic anemia. The number of RBCs decreases.
Hemorrhage: With active bleeding the number of RBCs decreases. It takes time (several hours), however, for the RBC count to fall. Only if the blood volume is replenished with fluid will the RBC count diminish.
Dietary deficiency: With certain vitamin or mineral deficiencies (e.g., iron, vitamin B12), the RBC size or number is decreased.
Bone marrow failure: This results in reduced synthesis of RBC.
Prosthetic valves: Prosthetic valves cause mechanical trauma to the RBC. The RBC survival time is shortened and numbers diminish.
Renal disease: Erythropoietin is made in the kidney and is a strong stimulant to RBC production. With reduced levels of erythropoietin, the RBC numbers diminish.
Normal pregnancy: Normally there is increased blood volume during pregnancy because of a chronic state of overhydration. Combined with a relative “malnourished” state, the RBC count per cubic millimeter of blood is diminished.
Rheumatoid/collagen-vascular diseases (e.g., rheumatoid arthritis, lupus, sarcoidosis): Chronic illnesses are associated with reduced production of RBCs.
Related Tests
Hematocrit (p. 277). This is a measurement of the percentage of the total blood volume taken up by the RBCs. It is closely associated with the hemoglobin value and the RBC count.
Hemoglobin (p. 281). This is a measurement of the concentration of hemoglobin in the blood. It is closely associated with the RBC count and hematocrit value.
Red Blood Cell Indices (see following test). These indices provide information about the size and hemoglobin content of the RBC.
Red Blood Cell Indices (RBC Indices, Mean Corpuscular Volume [MCV], Mean Corpuscular Hemoglobin [MCH], Mean Corpuscular Hemoglobin Concentration [MCHC], Blood Indices, Erythrocyte Indices, Red Blood Cell Distribution Width [RDW])
Indications
The RBC indices provide information about the size (MCV and RDW), hemoglobin content (MCH), and hemoglobin concentration (MCHC) of RBCs. This test is useful in classifying anemias.
Test Explanation
This test is routinely performed as part of an automated complete blood cell count. The results of the RBC, hematocrit, and hemoglobin tests (see pp. 439, 277, and 281, respectively) are necessary to calculate the RBC indices. When investigating anemia, it is helpful to categorize the anemia according to the RBC indices, as shown in Box 2-18. Cell size is indicated by the terms “normocytic,” “microcytic,” and “macrocytic.” Hemoglobin content is indicated by the terms “normochromic,” “hypochromic,” and “hyperchromic.” Additional information about the RBC size, shape, color, and intracellular structure is described in the blood smear study (see p. 710).
Mean Corpuscular Volume
The MCV is a measure of the average volume, or size, of a single RBC and is therefore used in classifying anemias. MCV is derived by dividing the hematocrit by the total RBC count:

Normal values vary according to age and gender. When the MCV value is increased, the RBC is said to be abnormally large, or macrocytic. This is most frequently seen in megaloblastic anemias (e.g., vitamin B12 or folic acid deficiency). When the MCV value is decreased, the RBC is said to be abnormally small, or microcytic. This is associated with iron-deficiency anemia or thalassemia. It is important to recognize that a significant number of patients with disorders associated with a variation in MCV may, in fact, not have an abnormality in MCV. For example, only 65% of patients with iron-deficiency anemia will have a reduced MCV. Furthermore, the normal values for MCV and all of the other RBC indices vary considerably. Each laboratory must develop its own normal index values.
Mean Corpuscular Hemoglobin
The MCH is a measure of the average amount of hemoglobin within an RBC. MCH is derived by dividing the total hemoglobin concentration by the number of RBCs:

Because macrocytic cells generally have more hemoglobin and microcytic cells have less hemoglobin, the causes for these values closely resemble those for the MCV value. This has been documented with the use of automated counting instruments. The MCH adds very little information to the other indices.
Mean Corpuscular Hemoglobin Concentration
The MCHC is a measure of the average concentration or percentage of hemoglobin within a single RBC. MCHC is derived by dividing the total hemoglobin concentration by the hematocrit:

When values are decreased, the cell has a deficiency of hemoglobin and is said to be hypochromic (frequently seen in iron-deficiency anemia and thalassemia). When values are normal, the anemia is said to be normochromic (e.g., hemolytic anemia). RBCs cannot be considered hyperchromic. Only 37 g/dL of hemoglobin can fit into the RBC. Alteration in RBC shape (spherocytosis, acute transfusion reactions, erythroblastosis fetalis) may cause automated counting machines to indicate MCHC levels above normal.
Red Blood Cell Distribution Width
The RDW is an indication of the variation in RBC size. It is calculated by a machine using the MCV and RBC values. Variations in the width of the RBCs may be helpful when classifying certain types of anemia. The RDW is essentially an indicator of the degree of anisocytosis, a blood condition characterized by RBCs of variable and abnormal size.
The newer electronic cell counting machines are able to sort out RBCs according to size and compare those sizes to a histogram. Normally all the RBCs are about the same size with very little variation. This creates a histogram with a single narrowed peak. Certain diseases change the size of some of the RBCs, whereas the less abnormal RBCs are less affected. For example, with folic acid deficiency or iron deficiency, the newer RBCs are more significantly affected than the older cells and therefore will be of significantly different size. This creates a histogram with multiple peaks indicating large numbers of cells at variable sizes.
Interfering Factors
• Abnormal RBC size may affect the MCH and MCHC.
• Extremely elevated WBC counts (>50,000) may increase the MCV and MCH indices when processed by automated counters.
• Large RBC precursors, for example, reticulocytes (see p. 452), cause an abnormally high MCV. This commonly occurs in response to anemias when the bone marrow is not pathologic.
• Marked elevation in lipid levels (>2000 mg/dL) causes automated cell counters to indicate high hemoglobin levels. MCV, MCHC, and MCH will be calculated falsely high.
• The presence of cold agglutinins also falsely elevates MCHC, MCH, and MCV.
Drugs that may increase MCV results include azathioprine, phenytoin, and zidovudine.
Test Results and Clinical Significance
Increased MCV
Pernicious anemia (vitamin B12 deficiency),
These are the most common causes of macrocytic anemia. These vitamin deficiencies may be caused by malnutrition, malabsorption, competitive parasites, or enzyme deficiencies that impair utilization of these vitamins.
Antimetabolite therapy: This form of chemotherapy for cancer treatment and, in lesser doses, for arthritis treatment, acts as vitamin B12 and folate inhibitors and can cause a macrocytic anemia.
Alcoholism: This is probably more related to malnutrition.
Chronic liver disease: The pathophysiology of this observation is multifactorial and includes poor nutrition, erythropoietin alterations, and the effects of chronic illness.
Increased MCHC
Spherocytosis: The automated cell counter's false perception of an elevation in the MCHC is caused by a variation in the shape of the RBC. The RBC can hold only 37 g/dL of hemoglobin. There can be no “real” hyperchromatism.
Intravascular hemolysis: This is caused by free hemoglobin in the blood. The automated counter sees the free hemoglobin and incorporates that into its calculations.
Cold agglutinins: Cold agglutinins cause the misperception of increased MCV and decreased hematocrit. The automated machine calculates a falsely high MCHC.
Increased RDW
B12 vitamin or folate-deficiency anemia:
Increased variation in RDW is caused by a combination of factors in these diseases. RBC fragmentation alters RBC size and shape. Furthermore, new cells produced when the deficiency was greatest will be markedly different in size and shape than the older RBCs that were produced before the deficiencies were as severe.
Hemoglobinopathies (e.g., sickle cell or C disease): Fragmentation increases RDW variation. Furthermore, different RBCs have different amounts of pathologic hemoglobin and therefore will be affected by fragmentation to varying degrees.
Hemolytic anemias: Fragmentation increases RDW variation.
Posthemorrhagic anemias: The marrow's response to bleeding is to release premature RBCs into the bloodstream. These are larger than mature RBCs and contribute to RDW variation.
Related Tests
Hematocrit (p. 277). This is a measurement of the percentage of the total blood volume taken up by the RBCs. It is closely associated with the hemoglobin value and the RBC count.
Hemoglobin (p. 281). This is a measurement of the concentration of hemoglobin in the blood. It is closely associated with the RBC count and hematocrit value.
Red Blood Cell (RBC) Count (p. 439). This is a measurement of the number of RBCs per cubic millimeter of blood. It is closely associated with the hemoglobin and hematocrit values.
Renin Assay, Plasma (Renin Activity, Plasma Renin Activity [PRA], Plasma Renin Concentration [PRC])
Indications
PRA is used to evaluate hypertension. It is helpful in the differential diagnosis of aldosteronism.
Test Explanation
Renin is an enzyme released by the juxtaglomerular apparatus of the kidney into the renal veins in response to hyperkalemia, sodium depletion, decreased renal blood perfusion, or hypovolemia. Renin activates the renin-angiotensin system, which produces angiotensins I, II, and III (p. 63), powerful vasoconstrictors that also stimulate aldosterone production from the adrenal cortex. Angiotensin and aldosterone increase the blood volume, blood pressure, and serum sodium (Figure 2-26). After release of renin from the kidney into the bloodstream, angiotensinogen, an alpha2 globulin that is made in the liver, is converted into angiotensin I. This is then converted into angiotensin II in the lung.

Renin is not actually measured in this test. Plasma renin activity (PRA) measures enzyme ability to convert angiotensinogen to angiotensin I and is limited by the availability of angiotensinogen. The PRA test actually measures, by radioimmunoassay, the rate of angiotensin I generation per unit time. This is a commonly used renin assay. The specimen must be drawn under ideal circumstances, handled by the local laboratory correctly, and transferred to the central laboratory in a timely manner. Even then, results may vary significantly.
The PRA is a screening procedure for the detection of essential, renal, or renovascular hypertension. The PRA may be supplemented by other tests, such as the renal vein renin assay. A determination of the PRA and a simultaneous measurement of the plasma aldosterone level are used in the differential diagnosis of primary versus secondary hyperaldosteronism (Table 2-44). Patients with primary hyperaldosteronism (adrenal adenoma overproducing aldosterone or Conn syndrome) will have increased aldosterone production associated with decreased renin activity. The aldosterone/renin ratio is ≥20.
TABLE 2-44
Differential Diagnosis Using Renin and Aldosterone Risk
| Disease | Renin (PRA) | Aldosterone |
| Conn syndrome | Low | High |
| Renal artery stenosis (or occlusion) | High | High |
| Primary renal disease | High | High |
| Increased salt intake | Low | Low |
| Salt restriction | High | High |
| Hypokalemia | Low | Low |
| Sodium-losing diuretic therapy | High | High |
| Addison disease | High | Low |
| Cushing syndrome | Low | High |
| Essential hypertension | Low | Normal |
Patients with secondary hyperaldosteronism (caused by renovascular occlusive disease or primary renal disease) will have increased levels of aldosterone and plasma renin.
Renal vein assays for renin are used to diagnose and lateralize renovascular hypertension, that is, hypertension that is related to inappropriately high renin levels from a diseased kidney or a hypoperfused kidney. The renal veins can be identified using injection of a radiopaque dye into the inferior vena cava. A catheter is placed into each renal vein, and blood is withdrawn from each vein. PRA is determined in each sample. If hypertension is caused by renal artery stenosis or renal pathology, the renal vein renin level of the affected kidney should be 1.5 or more times greater than that of the unaffected kidney or peripheral venous sample. If the levels are the same, the hypertension is not caused by a renovascular source. This is very helpful in determining whether a stenosis seen on a renal angiogram is significantly contributing to hypertension. Any stenosis identified on an arteriogram would not be considered severe enough to cause renin-related hypertension if renin levels from the renal vein were not at least 1.4 times those of the opposite kidney. Another cause for the patient's elevated blood pressure should be considered.
The renin stimulation test can be performed to more clearly diagnose and distinguish primary and secondary hyperaldosteronism. In this test, PRA is obtained while the patient is in the recumbent position and on a low-salt diet. The PRA is then repeated with the patient on the same diet while the patient is standing erect. In primary hyperaldosteronism the blood volume is greatly expanded. A change in position or reduced salt intake will not result in decreased renal perfusion or sodium level. Therefore renin levels do not increase. In secondary hyperaldosteronism (or normal persons with essential hypertension), the renal perfusion decreases while in the upright position and sodium levels decrease with decreased intake. Therefore renin levels increase.
The PRA is assessed as part of the captopril test (a screening test for renovascular hypertension). Patients with renovascular hypertension have greater falls in blood pressure and increases in PRA after administration of angiotensin-converting enzyme (ACE) inhibitors than do those with essential hypertension. For the captopril test, the patient receives an oral dose of captopril (ACE inhibitor) after a baseline PRA test, and blood pressure measurements are then taken. Subsequent blood pressure measurements and a repeat PRA test at 60 minutes are used for test interpretation. This is an excellent screening procedure to determine the need for a more invasive radiographic evaluation (such as digital subtraction renal arteriography [p. 988] or bilateral renal arteriography [p. 988]).
Potential Complications
• Allergic reactions to iodinated dye can occur during the renal vein renin assay. The reaction may vary from mild flushing, itching, and urticaria to severe, life-threatening anaphylaxis (evidenced by respiratory distress, drop in blood pressure, shock). In the unusual event of anaphylaxis, the patient may be treated with diphenhydramine (Benadryl), steroids, and epinephrine. Oxygen and endotracheal equipment should be on hand for immediate use.
Interfering Factors
• Renin is increased during pregnancy by virtue of increased substrate proteins concomitantly present in the serum during testing.
• Renin is increased with reduced salt intake. Reduced sodium acts as a direct stimulant to renin production.
• Renin is increased by ingestion of large amounts of licorice. Licorice has an aldosterone-like effect. This increases sodium reabsorption in the kidney and raises blood pressure, which in turn inhibits renin production.
• There is a diurnal variation in renin production. Values are higher early in the day.
• Renin levels are increased when the patient is in an upright position. Normally the upright position decreases renal perfusion because the blood pools in the veins of the lower extremities. This decreased renal perfusion is a strong stimulant to renin production. Renin levels are decreased in the recumbent position for the same reason (i.e., renal perfusion is increased in the recumbent position and renin levels diminish).
Spironolactone interferes with renin testing and should be discontinued 4 to 6 weeks before testing.
Drugs that increase levels of renin include ACE inhibitors, antihypertensives, diuretics, estrogens, oral contraceptives, and vasodilators.
Drugs that decrease renin levels include beta blockers, clonidine, licorice, NSAIDs, potassium, and reserpine.
 Clinical Priorities
Clinical Priorities
• There is a diurnal variation in renin production. Renin levels are higher in the morning. A morning blood sample is usually drawn.
• Renin levels are affected by body position. Levels are higher in the upright position and decreased in the recumbent position.
• Renin levels are increased with reduced salt intake, because reduced sodium levels are a stimulus to renin production.
Procedure and Patient Care
Before
Explain the procedure to the patient.
Instruct the patient to maintain a normal diet with a restricted amount of sodium (approximately 3 g/day) for 3 days before the test.
Instruct the patient to discontinue licorice and any medications that may interrupt renin activity for 2 to 4 weeks before the test as ordered by the physician.
• Plan to draw a morning (8:00 AM to 10 AM) sample, because renin values are higher in the morning.
For stimulation tests, instruct the patient to significantly reduce sodium intake (supplemented with potassium) for 3 days before testing.
During
• The test may be performed with the patient in an upright position.
• For the more commonly performed stimulation test, the blood is drawn in the recumbent and upright positions.
• Ensure that the patient stands or sits upright for 2 hours before the blood is drawn.
• If a recumbent sample is ordered, have the patient remain in bed in the morning until the blood sample has been obtained.
• It is best to release the tourniquet immediately before obtaining the blood specimen, because stasis can lower renin levels.
• Collect a venous blood sample and place it in a chilled lavender-top tube with ethylene diamine tetraacetic acid (EDTA) as an anticoagulant. Heparin can falsely decrease results.
• Gently invert the blood tube to allow adequate mixing of the blood sample and the anticoagulant.
• Record the patient's position, dietary status, and time of day on the laboratory slip.
• Place the tube of blood on ice, and immediately send it to the laboratory.
• In the laboratory, the blood will be centrifuged and the serum frozen.
Test Results and Clinical Significance
Increased Levels
Essential hypertension: A small percentage of these patients have renin hypertension.
Malignant hypertension: A large percentage of these patients with aggressive hypertensive episodes have secondary hyperaldosteronism (usually because of renal vascular occlusion or stenosis).
Renovascular hypertension: Renal artery stenosis or occlusion decreases the renal blood flow, which is a strong stimulant to renin production.
Chronic renal failure: Diseases of the kidney can stimulate the production of renin.
Salt-losing GI disease (vomiting or diarrhea): These patients develop hyponatremia, which is a strong stimulant to renin production.
Addison disease: These patients are hyponatremic, which is a strong stimulant to renin production.
Renin-producing renal tumor: Tumors of the juxtaglomerular apparatus are rare. They can produce renin.
Bartter syndrome: This syndrome is associated with potassium wasting in the kidney, high renin levels, and high aldosterone levels. This is caused by a tubular defect in sodium reabsorption.
Cirrhosis: These patients have increased total body water, which dilutes sodium. Sodium levels are chronically low, which is a stimulant for renin production.
Hyperkalemia: This is a direct stimulant for renin production.
Hemorrhage/hypovolemia: Any form of hypotension (including cardiogenic or septic shock) is associated with a reduction in the renal blood flow, which is a strong stimulant to renin production.
Decreased Levels
Primary hyperaldosteronism: This is usually caused by an adrenal adenoma, and aldosterone levels are high. Aldosterone inhibits further renin production.
Steroid therapy: Glucocorticosteroids also have an aldosterone effect, which acts to increase serum sodium levels, decrease potassium levels, and increase blood volume. These responses all tend to diminish renin levels.
Congenital adrenal hyperplasia: An enzyme defect in cortisol synthesis causes an accumulation of cortisol precursors, some of which have strong aldosterone-like activity. These act to increase serum sodium levels, decrease potassium levels, and increase blood volume, all of which tend to diminish renin levels.
Reticulocyte Count (Retic Count)
Indications
The reticulocyte count is an indication of the ability of the bone marrow to respond to anemia and make RBCs. It is used to classify and monitor therapy of anemias.
Test Explanation
The reticulocyte count is a test for determining bone marrow function and evaluating erythropoietic activity. This test is also useful in classifying anemias. A reticulocyte is an immature red blood cell (RBC) that can be readily identified under a microscope by staining the peripheral blood smear with Wright or Giemsa stain. It is an RBC that still has some microsomal and ribosomal material left in the cytoplasm. It sometimes takes a few days for that material to be cleared from the cell. Normally there are a small number of reticulocytes in the bloodstream.
The reticulocyte count gives an indication of RBC production by the bone marrow. Increased reticulocyte counts indicate the marrow is releasing an increased number of RBCs into the bloodstream, usually in response to anemia. A normal or low reticulocyte count in a patient with anemia indicates that the marrow response to the anemia by way of production of RBCs is inadequate and perhaps is contributing to or is the cause of the anemia (as in aplastic anemia, iron deficiency, vitamin B12 deficiency, depletion of iron stores). An elevated reticulocyte count found in patients with a normal hemogram indicates increased RBC production compensating for an ongoing loss of RBCs (hemolysis or hemorrhage).
Because the reticulocyte count is a percentage of the total number of RBCs, a normal to low number of reticulocytes can appear high in the anemic patient, because the total number of mature RBCs is low. To determine if a reticulocyte count indicates an appropriate erythropoietic (RBC marrow) response in patients with anemia and a decreased hematocrit, the reticulocyte index is calculated as follows:
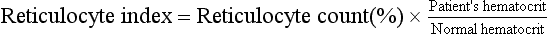
The reticulocyte index in a patient with a good marrow response to the anemia should be 1.0. If it is below 1.0, even though the reticulocyte count is elevated, the bone marrow response is inadequate in its ability to compensate (as seen in iron deficiency, vitamin B12 deficiency, marrow failure). In these clinical situations, if iron or vitamin B12 is administered, the reticulocyte count will rise significantly to the point that the index equals or exceeds 1.0.
Interfering Factors
• Pregnancy may cause an increased reticulocyte count.
• Howell-Jolly bodies are blue stippling material in the RBC that occurs in severe anemia or hemolytic anemia. The RBCs containing these Howell-Jolly bodies look like reticulocytes and can be miscounted by some automated counter machines as reticulocytes; this gives a falsely high number of reticulocytes.
Test Results and Clinical Significance
Increased Levels
Hemolytic anemia (e.g., immune hemolytic anemia, hemoglobinopathies, hypersplenism, trauma from a prosthetic heart valve): The RBC survival is decreased and RBCs are destroyed at a faster rate than normal. The marrow attempts to compensate for the shortened RBC survival by producing large numbers of RBCs, some of which are immature RBCs called reticulocytes.
Hemorrhage (3 to 4 days later): In response to significant blood loss, the marrow attempts to compensate by producing large numbers of RBCs, some of which are immature RBCs called reticulocytes.
Hemolytic disease of the newborn: Immune-mediated destruction of RBCs reduces RBC survival. The marrow attempts to compensate for the shortened RBC survival by producing large numbers of RBCs, some of which are immature RBCs called reticulocytes.
Treatment for iron, vitamin B12, or folate deficiency: After replacement treatment for anemia caused by nutritional deficiency, the marrow responds by increasing production of RBCs, some of which are immature RBCs called reticulocytes.
Rheumatoid Factor (RF, Rheumatoid Arthritis [RA] Factor)
Test Explanation
RA is a chronic inflammatory disease that affects most joints, especially the metacarpal and phalangeal joints, the proximal interphalangeal joints, and the wrists; however, any synovial joint can be involved. The American College of Rheumatology has defined criteria for the diagnosis of RA. They include:
• Morning stiffness for at least 6 weeks
• Pain in at least one joint for the preceding 6 weeks
• Swelling in at least one joint for the preceding 6 weeks
• Symmetric bilateral joint swelling
In this disease, abnormal immunoglobulin (Ig) G antibodies produced by lymphocytes in the synovial membranes act as “antigens.” Other IgG and IgM antibodies in the patient's serum react with the fc component of the abnormal synovial antigenic IgG to produce immune complexes. These immune complexes activate the complement system and other inflammatory systems to cause joint damage. The reactive IgM and sometimes IgG and IgA make up what is called the RF. IgG and IgA can also react to the synovial “IgG antigen.” Tissues other than the joints, including blood vessels, lungs, nerves, and heart, may also be involved in the autoimmune inflammation.
Tests for RF are directed toward identification of the IgM antibodies. The exact role, if any, that RF plays in the pathophysiology of the disease is not well known. Approximately 80% of patients with RA have positive RF titers. To be considered positive, RF must be found in a dilution of greater than 1:80; when RF is found in titers of less than 1:80, diseases such as systemic lupus erythematosus (SLE), scleroderma, and other autoimmune conditions should be considered. Although the normal value is “no rheumatoid factor identifiable at low titers,” a small number of normal patients will have RF present in a very low titer. Furthermore, a negative RF does not exclude the diagnosis of RA. When the nephelometric testing procedure is used, the normal value is considered to be less than 60 units/mL. RF is not a useful disease marker, because it does not disappear in patients who are experiencing a remission of symptoms.
There are many serologic methods for detecting RF. The sheep cell agglutination test or the latex fixation test was most easily performed in the past. Better quantitation is now obtained by nephelometry. In the sheep cell agglutination test, rabbit IgG is placed on the sheep red blood cells (RBCs). When this is mixed with the patient's serum (which has been serially diluted), visual agglutination occurs if any RF is present. In the latex fixation test, human IgG is placed on a synthetic latex particle and mixed with the patient's serum. Visual agglutination is then detected if RF is present (Figure 2-27).

Other autoimmune diseases (see Table 2-5 on p. 90), such as SLE or Sjögren syndrome, also may cause a positive RF test. RF is occasionally seen in patients with tuberculosis, chronic hepatitis, infectious mononucleosis, and subacute bacterial endocarditis as well.
Ribosome P Antibodies (Ribosomal P Ab, Anti-Ribosome P Antibodies)
Indications
Ribosome P antibodies are used as an adjunct in the evaluation of patients with lupus erythematosus (LE).
Test Explanation
This antibody test should not be confused with anti-extractable nuclear antibodies (antiribonucleoprotein antibody, p. 79). Antibodies to ribosome P proteins are considered highly specific for LE, and have been reported in patients with central nervous system (CNS) involvement (i.e., lupus psychosis). This antibody is therefore an aid in the differential diagnosis of neuropsychiatric symptoms in patients with LE. Because patients with LE may manifest signs and symptoms of CNS diseases including neuropsychiatric symptoms, the presence of antibodies to ribosome P protein may be useful in the differential diagnosis of such patients. Most patients with LE do not have detectable levels of antibodies to ribosome P protein. But when they do, CNS involvement should be considered. This test is performed using immunofluorescent antibodies.
Rubella Antibody (German Measles, Hemagglutination Inhibition [HAI])
Normal Findings
| Method | Result | Interpretation |
| HAI | <1:8 | No immunity to rubella |
| HAI | >1:20 | Immunity to rubella |
| Latex agglutination (LA) | Negative | No immunity to rubella |
| Enzyme-linked immunosorbent assay (ELISA) IgM | <0:9 international units/mL | No infection |
| ELISA IgM | >1.1 international units/mL | Active infection |
| ELISA IgG | <7 international units/mL | No immunity to rubella |
| ELISA IgG | >10 international units/mL | Immunity to rubella |
Indications
Screening for rubella antibodies is performed to detect immunity to rubella (the causative agent for German measles). This is important for pregnant women or health care providers working with pregnant women. It is also used to diagnose rubella in newborns, children, and adults.
Test Explanation
These tests detect the presence of IgG and/or IgM antibodies to rubella. They become elevated a few days to a few weeks (depending on the method of testing) after the onset of the rash. IgM tends to disappear after about 6 weeks. IgG, however, persists at low but detectable levels for years (Table 2-45).
TABLE 2-45
| Indication | Antibody |
| Evaluate immune status | IgG |
| Identify active infection | IgM or IgG, acute and convalescent |
| Identify congenital infection | IgM |
These antibodies become elevated in patients with active rubella infection or with past infections. In the past decade, children have been vaccinated with rubella to prevent the effects of the disease and to minimize infection. Rubella testing documents immunity to rubella. Rubella immunity testing is suggested for all health care workers. Most importantly, however, it is done to verify the presence or absence of rubella immunity in pregnant women, because congenital rubella infection in the first trimester of pregnancy is associated with congenital abnormalities (heart defects, brain damage, deafness), abortion, or stillbirth.
The term TORCH (toxoplasmosis, other, rubella, cytomegalovirus, herpes) has been applied to infections with recognized detrimental effects on the fetus. The effects on the fetus may be direct or indirect (e.g., precipitating abortion or premature labor). Included in the category of “other” are infections (e.g., syphilis). All of these tests are discussed separately.
If the woman's titer is greater than 1:10 to 1:20, she is not susceptible to rubella. If the woman's titer is 1:8 or less, she has little or no immunity to rubella. Pregnant women should be strongly advised to stay away from any small children, especially those with symptoms of an upper respiratory tract infection (prodromal symptoms of rubella). In addition, all health care personnel associated with maternal and child care should be screened for rubella. Immunization, if required, is not done during pregnancy but should be done before pregnancy or after delivery for nonimmune women.
A change in the HAI titer (measures IgG and IgM) from the acute to the chronic phase in a patient with a rash is the most useful method of demonstrating that the rash was related to a rubella infection. With a rubella rash, diagnosis of rubella is confirmed by obtaining an acute sample (approximately 3 days after the onset of the rash) and a convalescent sample (approximately 2 to 3 weeks later). A fourfold increase in the acute to the convalescent titer indicates that the rash was caused by an active rubella infection. Alternatively, in a pregnant woman with a rash suspected to be from rubella, an IgM antibody titer can be measured. If the titer is positive, recent infection has occurred. IgM titers appear 1 to 2 days after onset of the rash and disappear 5 to 6 weeks after infection.
Antirubella antibody testing is also used to diagnose rubella in infants (congenital rubella). Rubella is suspected in low-birth-weight (LBW) infants. Although IgG antibodies can be passed from mother to fetus, IgM antirubella antibodies cannot pass through the placenta. If an infant has IgM antibodies, acute congenital or newborn rubella is suspected. Antibody testing is often used in children with congenital abnormalities that may have resulted from congenital rubella infection. This test is also recommended for anyone with a rash that may be related to rubella.
The HAI method tests for IgG and IgM. LA detects IgG only and is often used as a simple screen for immunity. Enzyme-linked immunosorbent assay (ELISA) methods for detecting IgG and IgM are now the standard for rubella testing. They provide a more accurate testing method and antibody quantities can be determined.
Rubeola Antibody
Indications
This test is used to diagnose rubeola infection (measles). It is more commonly used, today, to document immunity to infection by prior vaccination or clinical disease.
Test Explanation
Rubeola is a RNA paramyxovirus that is known to cause the measles (not German measles—see Rubella). Upper respiratory symptoms, fever, conjunctivitis, a rash, and Koplik spots on the buccal mucosa highlight the disease. Since the 1970s, children have been vaccinated to prevent this disease. Although it is usually a self-limiting disease, the virus can easily be spread (by respiratory droplets) to nonimmune pregnant women and cause preterm delivery or spontaneous abortion.
Testing for rubeola includes indirect immunofluorescence serologic identification of IgG and IgM antibodies. The first represents a previous infection. The latter indicates an acute infection. A fourfold rise in IgM indicates a current infection.
This test is used to diagnose measles in patients with a rash or viral syndrome when the diagnosis cannot be made clinically. Even more importantly, however, this test is used to establish and document immunity (active—by previous measles infection, or passive—by previous vaccination). Populations commonly tested to document immunity include college students, health care workers, and pregnant women.
Test Results and Clinical Significance
Active rubeola infection: These patients may not have the “classic” clinical signs of measles and diagnosis can be made with certainty through the identification of IgM antibodies in the patient's serum.
Previous rubeola infection leading to immunity: These patients have IgG antibodies but do not have IgM antibodies. They are protected from the disease because of previous active infection or vaccination.
Related Test
Rubella Antibody (p. 457). This test is used to diagnose German measles and to document immunity to the same.
Septin 9 DNA Methylation Assay (Methylated Septin 9, mSEPT9, ColoVantage)
Indications
This test is used to screen asymptomatic patients for colorectal cancer. Its use as a screening modality has not been established, but its main benefit may be in the early detection of colorectal cancer in patients who refuse colonoscopy or stool testing.
Test Explanation
Because of the inconvenience and discomfort associated with routine colorectal cancer screening (see colonoscopy, p. 591; stool for occult blood testing, p. 857), about half of Americans 50 to 75 years old do not follow recommended colorectal cancer (CRC) screening guidelines, leaving 40 million individuals unscreened. This precludes the opportunity for the early detection of an intestinal cancer. Recently a blood test for the detection of methylated DNA from the septin 9 (SEPT9) gene has been developed that, when positive, is very sensitive for the presence of a colorectal cancer. Using real-time methylated PCR, septin can be isolated and quantified from extracted nucleic acid in the plasma. This test has been validated in several clinical studies and shows a strong association between detection of mSEPT9 in blood plasma and the presence of colorectal cancer. Although more expensive than stool for occult blood testing, this real-time PCR laboratory blood test outperforms the stool test without the unpleasantness of a stool collection and may improve compliance for screening for colorectal cancer. Although the SEPT9 methylated DNA test may perform comparably to colonoscopy in detecting CRCs, it lacks the advantage of being potentially able to remove any precancerous polyps, thereby decreasing subsequent risks of cancer. Furthermore, Sept9 does not perform well for adenoma detection.
A positive test result means that there is an increased likelihood for the presence of a colorectal cancer or polyp. Individuals with positive test results are encouraged to undergo a diagnostic colonoscopy. Not all individuals with colorectal cancer will have a positive test result. Therefore individuals with a negative result should follow usual colorectal cancer screening guidelines.
Test Results and Clinical Significance
Increased Levels
Although it is clear that CRC screening reduces mortality by detecting the disease in its earliest stages when it is most effectively treated, only one half of Americans age 50 and older currently undergo any kind of screening. Reasons for not complying with colonoscopy include the time-consuming nature of the procedure and concern about invasiveness. In addition to the challenges of patient compliance with stool testing, such as the requirement for multiple samples and the handling of specimens, the performance of these tests is quite variable. Newer stool-based tests such as the immunochemical FOBT (FIT), have demonstrated sensitivity for adenoma detection.
Related Tests
Colonoscopy (p. 591). This is an endoscopic study of the entire colon and rectum that is the most effective screening study for the detection of early colorectal cancer.
Stool for Occult Blood (p. 857). Testing the stool for occult blood or DNA is an alternative accurate method of screening for early colorectal cancer.
Apt Test (p. 848). This is a method of identifying blood in newborn stool and differentiating the newborn's from the mother's blood.
Serotonin (5-Hydroxytryptamine, 5-HT) and Chromogranin A
Indications
This test is used in conjunction with, or as an alternative to, 5-HIAA (p. 928) or serum chromogranin A measurements as a first-line test in the diagnosis of carcinoid syndrome or symptoms such as flushing. It is also used to monitor patients with known or treated carcinoid tumors.
Test Explanation
Serotonin is synthesized from the essential amino acid tryptophan chiefly in the gastrointestinal enterochromaffin cells (EC-cells). Many different stimuli can release serotonin from EC-cells. After it is secreted, in concert with other gut hormones, serotonin increases GI blood flow, motility, and fluid secretion. On first pass through the liver, 30% to 80% of serotonin is metabolized, predominantly to 5-hydroxyindoleacetic acid (5-HIAA), which is then excreted by the kidneys.
The main diseases that may be associated with measurable increases in serotonin are neuroectodermal tumors, in particular tumors arising from EC-cells. These tumors are collectively referred as carcinoids. They are subdivided into foregut carcinoids, arising from respiratory tract, stomach, pancreas, or duodenum (approximately 15% of cases); midgut carcinoids, occurring in the jejunum, ileum, or appendix (approximately 70% of cases); and hindgut carcinoids, which are found in the colon or rectum (approximately 15% of cases). In patients with more advanced tumors, serotonin is elevated in nearly all patients with midgut tumors, but only in approximately 50% of those with foregut carcinoids, and in no more than 20% of individuals with hindgut tumors. Foregut and hindgut tumors often have low or absent serotonin.
Carcinoids display a spectrum of aggressiveness with no clear distinguishing line between benign and malignant. The majority of carcinoid tumors do not cause significant clinical symptoms. Most symptoms are caused by elevated serotonins (carcinoid syndrome). The carcinoid syndrome consists of flushing, diarrhea, right-sided valvular heart lesions, and bronchoconstriction. The carcinoid syndrome is usually caused by midgut tumors. Because midgut tumors drain into the liver, nearly all of the serotonin is metabolized on first pass. Carcinoid symptoms, therefore, do not usually occur until liver or other distant metastases have developed that bypass the hepatic metabolism.
Diagnosis of carcinoid tumors with symptoms suggestive of carcinoid syndrome rests on measurements of serum serotonin, urinary 5-HIAA (p. 928), and serum chromogranin A (a peptide that is cosecreted alongside serotonin by the neuroectodermal cells). Metastasizing midgut carcinoid tumors usually produce blood or serum serotonin concentrations greater than 1,000 ng/mL. Only a minority of patients with carcinoid tumors will have elevated serotonin blood levels because the liver rapidly metabolizes the serotonin. It is usually impossible to diagnose small carcinoid tumors (>95% of cases) without any symptoms suggestive of carcinoid syndrome by measurement of serotonin, 5-HIAA, or chromogranin A. It is only after carcinoid tumors metastasize that serotonins become detectable because the blood that drains the metastatic carcinoid tumors carries serotonin from the metastatic tumors but does not pass through the liver for metabolism. In most cases, if a person has true carcinoid syndrome symptoms, serotonin levels are significantly elevated. If none of three analytes are elevated, carcinoids can be excluded as a cause of those symptoms.
Disease progression can be monitored in patients with serotonin-producing carcinoid tumors by measurement of serotonin or chromogranin A in the blood. However, at levels greater than approximately 5000 ng/mL, there is no longer a linear relationship between tumor burden and blood serotonin levels. Urinary 5-HIAA and serum chromogranin A continue to increase in proportion to the tumor burden.
Chromogranin A also acts as a useful diagnostic marker for other neuroendocrine neoplasms, including carcinoids, pheochromocytomas, neuroblastomas, medullary thyroid carcinomas, some pituitary tumors, functioning and nonfunctioning islet-cell tumors, and other amine precursor uptake and decarboxylation (APUD) tumors. It can also serve as a sensitive means for detecting residual or recurrent disease in treated patients. Carcinoid tumors, in particular colon and rectal carcinoids, almost always secrete chromogranin A. Other neuroendocrine tumors, such as small cell carcinoma of the lung or prostate carcinoma, may also display elevated chromogranin A levels.
After being extracted from the serum by reversed-phase solid-phase extraction, serotonin is analyzed using liquid chromatography/tandem mass spectrometry and quantified using a stable isotope-labeled internal standard. Chromogranin A is measured in a homogeneous automated immunofluorescent assay. This assay uses technology based on time-resolved amplified cryptae emission.
Interfering Factors
Drugs that may cause increased serotonin levels include lithium, MAO-inhibitors, methyldopa, morphine, and reserpine.
Drugs that may decrease serotonin levels include selective serotonin reuptake inhibitors (e.g., fluoxetine).
Drugs that may cause increased chromogranin A levels include proton pump inhibitors (e.g., omeprazole) and should be discontinued 2 weeks before testing.
Procedure and Patient Care
During
• Collect venous blood in a red-top tube and deliver to the laboratory as soon as possible. Because most circulating 5-HT is contained in platelets, the preferred specimens for measurement either include all or most of the platelets (i.e., whole blood and platelet-rich plasma) or consist of serum from completely clotted specimens, a process that releases nearly all 5-HT from platelets.
• Note that testing is usually performed at a reference laboratory.
Related Tests
5-Hydroxyindoleacetic Acid (p. 928). This urinary test measures the quantity of 5-hydroxyindoleacetic acid, a metabolite of serotonin that is excreted in the urine. Its use is similar to serotonin and chromogranin A.
Sickle Cell Screen (Sickledex, Hemoglobin S [Hgb S])
Test Explanation
Both sickle cell disease (homozygous for Hgb S) and sickle cell trait (heterozygous for Hgb S) can be detected by this screening study. Sickle cell anemia results from a genetic homozygous defect and is caused by the presence of Hgb S instead of Hgb A (Figure 2-28). When Hgb S becomes deoxygenated, it tends to bend in a way that causes the red blood cells (RBCs) to assume a sickle shape. These sickled RBCs cannot pass freely through the capillaries, thus they cause plugging of the microvascular tree. This may compromise the blood supply to various organs. Hgb S is found in varying quantities in 8% to 10% of the black population.
Figure 2-28 Sickle cell anemia. Sickle cell hemoglobin (Hgb) is produced by a recessive allele of the gene encoding the beta chain of Hgb. It represents a single amino acid change from a glutamic acid to valine at the sixth position in the chain. In the folded beta chain the sixth position contacts the alpha chain, and the amino acid change causes the hemoglobin to aggregate into long chains, altering the shape of the cell.
The Sickledex test is a blood test that is positive (turbid or cloudy test fluid) if greater than 10% of the hemoglobin is Hgb S. This is only a screening test, and its sensitivity varies according to the method used by the laboratory. Double heterozygosity for sickle trait when combined with another hemoglobinopathy (e.g., Hgb C disease) can cause a sickling disease. The definitive diagnosis of sickle cell disease or trait is made by Hgb electrophoresis (p. 284) or high-performance liquid chromatography, in which Hgb S can be identified and quantified. Immunoassay methods using monoclonal antibodies are also being used to quantify Hgb S.
Because sickle cell and Hgb C and E diseases are all associated with genetic abnormalities that affect the Beta globin gene (HBB), PCR Beta globin gene testing can now be performed on amniotic fluid, thereby identifying the disease in the fetal state.
Interfering Factors
• Any blood transfusions within 3 to 4 months before the sickle cell test may cause false-negative results, because the donor's normal Hgb may dilute the recipient's abnormal Hgb S.
• Polycythemia may cause false-negative results.
• Infants less than 3 months of age may have false-negative results, because even infants with sickle cell disease have a significant amount of Hgb F in their RBCs at that age. Hgb F will not cause sickling. After 6 months of age the Hgb S variant increases in numbers in these infants. It is then that the test will be positive.
Drugs that may cause false-negative results include phenothiazines.
Procedure and Patient Care
After
• Apply pressure or a pressure dressing to the venipuncture site.
• Check the venipuncture site for bleeding.
• If the test is positive, Hgb electrophoresis should be performed.
• If the test is positive, genetic counseling should follow.
Inform patients with sickle cell anemia that they should avoid situations in which hypoxia may occur (e.g., strenuous exercise, air travel in unpressurized aircraft, travel to high-altitude regions).
Related Test
Hemoglobin Electrophoresis (p. 284). This test can identify and measure Hgb S. Sickle cell disease can be differentiated from the trait.
Sodium, Blood (Na)
Indications
This test is a part of the routine laboratory evaluation of most patients. It is one of the tests automatically performed when “serum electrolytes” are requested. This test is used to evaluate and monitor fluid and electrolyte balance and therapy.
Test Explanation
Sodium is the major cation in the extracellular space, in which there are serum levels of approximately 140 mEq/L. The concentration of sodium intracellularly is only 5 mEq/L. Therefore sodium salts are the major determinants of extracellular osmolality. The sodium content in the blood is a result of a balance between dietary sodium intake and renal excretion. Nonrenal (e.g., sweat) sodium losses normally are minimal.
Many factors regulate sodium balance. Aldosterone causes conservation of sodium by stimulating the kidneys to reabsorb sodium and decreasing renal losses. Natriuretic hormone, or third factor, is stimulated by increased sodium levels. This hormone decreases renal absorption and increases renal losses of sodium. Antidiuretic hormone (ADH), which controls the reabsorption of water at the distal tubules of the kidney, affects sodium serum levels by dilution or concentration.
Physiologically, water and sodium are closely interrelated. As free body water is increased, serum sodium is diluted and the concentration may decrease. The kidney compensates by conserving sodium and excreting water. If free body water were to decrease, the serum sodium concentration would rise; the kidney would then respond by conserving free water. Aldosterone, ADH (vasopressin), and natriuretic factor all assist in these compensatory actions of the kidney to maintain appropriate levels of free water.
An average dietary intake of approximately 90 to 250 mEq/day is needed to maintain sodium balance in adults. Symptoms of hyponatremia may begin when sodium levels are below 125 mEq/L. The first symptom is weakness. When sodium levels fall below 115 mEq/L, confusion and lethargy occur and may progress to stupor and coma if levels continue to decline. Symptoms of hypernatremia include dry mucous membranes, thirst, agitation, restlessness, hyperreflexia, mania, and convulsions.
Interfering Factors
• Recent trauma, surgery, or shock may cause increased levels because renal blood flow is decreased. Renin and angiotensin stimulate the secretion of aldosterone, which stimulates increased renal absorption of sodium.
Drugs that may cause increased levels include anabolic steroids, antibiotics, carbenicillin, clonidine, corticosteroids, cough medicines, estrogens, laxatives, methyldopa, and oral contraceptives.
Drugs that may cause decreased levels include angiotensin-converting enzyme (ACE) inhibitors, captopril, carbamazepine, diuretics, haloperidol, heparin, nonsteroidal antiinflammatory drugs, sodium-free intravenous (IV) fluids, sulfonylureas, triamterene, tricyclic antidepressants, and vasopressin.
Test Results and Clinical Significance
Increased Levels (Hypernatremia)
Increased Sodium Intake
Increased dietary intake: If sodium (usually in the form of salt) is ingested at high quantities without adequate free water, hypernatremia will occur.
Excessive sodium in IV fluids: The normal kidney can excrete about 450 to 500 mEq of sodium per day. If intake of sodium exceeds that amount in a patient without ongoing losses or a prior sodium deficit, sodium levels can be expected to rise.
Excessive Free Body Water Loss
Gastrointestinal (GI) loss (without rehydration): If free water is lost, residual sodium becomes more concentrated.
Excessive sweating: Although sweat does contain some sodium, most is free water. This causes the serum sodium to become more concentrated. If the water loss is replaced without any sodium, sodium dilution and hyponatremia can occur.
Extensive thermal burns: If the burn is extensive, serum and a great amount of free water are lost through the open wounds. Sodium becomes more concentrated. As fluid is replaced and the body physiologically responds by stimulating ADH, sodium can be diluted and hyponatremia may occur.
Diabetes insipidus: The deficiency of ADH and the inability of the kidney to respond to ADH causes large free water losses. Sodium becomes concentrated.
Osmotic diuresis: With osmotic diuresis (excluding hyperglycemia, see below), water may be lost at a rate that exceeds sodium loss. In those situations, sodium levels increase as a result of greater concentration. If, however, free water is therapeutically provided, sodium levels may become dilute and hyponatremia may occur.
Decreased Levels (Hyponatremia)
Increased Sodium Loss
Addison disease: Aldosterone and corticosteroid hormone levels are inadequate. Sodium is not reabsorbed by the kidneys and is lost in the urine.
Diarrhea, vomiting, or nasogastric aspiration: Sodium in the GI contents is lost with the fluid. Hyponatremia is magnified if IV fluid replacement does not contain adequate amounts of sodium.
Intraluminal bowel loss (ileus, mechanical obstruction): Great amounts of extracellular fluid are “third spaced” into the lumen of the dilated bowel. This fluid contains sodium. Hyponatremia is magnified if IV fluid replacement does not contain adequate amounts of sodium.
Diuretic administration: Many diuretics work by inhibiting sodium reabsorption by the kidney. Sodium levels can diminish.
Chronic renal insufficiency: The kidney loses its reabsorptive capabilities. Large quantities of sodium are lost in the urine.
Large-volume aspiration of pleural or peritoneal fluid: Sodium concentration is the same as serum in these fluids. The aspiration of these fluids is compensated by secretion of ADH, which acts to increase renal absorption of free water. Sodium becomes diluted.
Increased Free Body Water
Excessive oral water intake: Psychogenic polydipsia can dilute sodium.
Hyperglycemia: Each 60 mg/100 mL increase of glucose above normal decreases the sodium 1 mEq/L, because the osmotic effect of the glucose pulls in free water from the extracellular space and dilutes sodium. Also, sodium ketotic salts are lost in the urine. Sodium levels diminish further.
Excessive IV water intake: When IV therapy provides less sodium than maintenance and ongoing losses, sodium will be diluted. If sodium-free IV therapy is given to a patient who has a significant sodium deficit, sodium dilution will occur with rehydration.
Intraluminal bowel loss (ileus or mechanical obstruction):
Syndrome of inappropriate or ectopic secretion of ADH: Oversecretion of ADH stimulates the kidney to reabsorb free water. Sodium is diluted.
Related Tests
Sodium, Urine (p. 946). This measurement of sodium in the urine is helpful in assessing sodium and water balance.
Aldosterone (p. 43). More than any other hormone, aldosterone has a significant effect on sodium blood levels.
Antidiuretic Hormone (ADH) (p. 73). By affecting free body water excretion, sodium levels become diluted or concentrated.
Squamous Cell Carcinoma Antigen (SCC Antigen)
Indications
This test is used to determine the stage and prognosis of squamous cell carcinomas. It is also used to monitor the treatment of carcinomas and a variety of nonmalignant conditions.
Test Explanation
Squamous cell carcinoma (SCC) antigen is a glycoprotein that is expressed in normal epithelium and epithelial tissues. Although the neutral forms of SCC normally remain inside the cell, acidic SCC antigen is released and often elevated in patients who have squamous cell carcinomas or other nonmalignant squamous cell lesions. It can occur in several cancers (e.g., uterine, cervical, oral cavity, esophageal, lung, anal canal, skin). SCC antigen may be involved in the malignant behavior of squamous cell cancers. Consequently, serum concentrations of SCC antigen can be used to monitor various SCCs after surgical removal. Concentrations that remain persistently elevated or begin to increase following tumor removal suggest persistent or recurrent disease. There may be an association between serum SCC antigen concentrations and tumor stage, size, and tumor aggressiveness.
A variety of nonmalignant benign diseases of the skin (e.g., eczema, erythrodermic epidermitis, pemphigus, and psoriasis), lungs (e.g., TB, adult respiratory distress syndrome, sarcoidosis, presence of pleural effusion), and other common conditions may result in increased serum concentrations of SCC antigen. Thus, SCC antigen results alone should not be interpreted as evidence of the presence or absence of malignant disease.
Related Test
Carcinoembryonic Antigen (p. 145). This is a tumor marker used in evaluating several cancers.
Streptococcus Serologic Testing (Antistreptolysin O Titer [ASO], Antideoxyribonuclease-B Titer, [Anti-DNase – B, ADB], Streptococcus Group B Antigen Detection, Streptozyme)
Normal Findings
Test Explanation
Infections by group A Streptococcus are unique because they can be followed by a serious nonpurulent complication (such as rheumatic fever, scarlet fever, or glomerulonephritis). Serologic tests are used primarily to determine if a previous group A Streptococcus infection (pharyngitis, pyodermia, or pneumonia) has caused a poststreptococcal disease. These poststreptococcal diseases occur following the infection and after a period of latency during which the patient is asymptomatic. The latency period for glomerulonephritis is approximately 10 days, and for rheumatic fever is about 20 days.
These antibodies are directed against streptococcal extracellular products that are primarily enzymatic proteins. Serial rising titers of these antibodies over several weeks, followed by a slow fall in titers, are more supportive of the diagnosis of a previous streptococcal infection than is a single titer. The highest incidence of positive results is during the third week after the onset of acute symptoms of the poststreptococcal disease. By 6 months, only about 30% of patients have abnormal titers. By 12 months, levels return to normal.
One such extracellular enzyme produced by streptococcus is called streptolysin O, which has the ability to destroy (lyse) red blood corpuscles. The streptolysin O is antigenic stimulating the immunologic production of a neutralizing ASO antibody. ASO appears in the serum 1 week to 1 month after the onset of a streptococcal infection. A high ASO titer is not specific for a certain type of poststreptococcal disease (i.e., rheumatic fever versus glomerulonephritis), but merely indicates that a streptococcal infection is or has been present.
Like the ASO titer, ADB is used to detect previous streptococcal infections. Because a significant portion of individuals with normal antibody titers for one test will have elevated antibody titers for another test, one test is not used alone in the evaluation of streptococcal infections. The percentage of false-negatives can be reduced by performing two or more antibody tests. ADB is often run concurrently with the ASO titer and other serologic tests to provide more accurate results.
The Streptozyme assay detects antibodies to multiple extracellular antigens of group A Streptococcus, including antistreptolysin O, antistreptokinase, and antihyaluronidase. Approximately 80% of specimens positive by Streptozyme have antistreptolysin O, and 10% have antistreptokinase and/or antihyaluronidase. The remaining 10% of positive samples are apparently the result of ADB antibodies or other streptococcal extracellular antigens. Nephelometry is the laboratory method used to identify most of these antibodies.
Streptococcus Group B antigens accumulate in CSF, serum, or urine and provide a direct qualitative detection of bacterial antigens. These antigens indicate acute infection and are not related to post streptococcal sequelae as described. Confirmatory diagnosis of streptococcal infection is done by cultures (p. 761). Samples with extremely low levels of antigen may yield negative results.
Rapid antigen detection (strept screen) testing is another immunologic test in which the Streptococcus organism can be identified directly from the swab specimen. The rapid serologic tests can be performed in about 15 minutes in any lab or in most physicians' offices that treats children. This test is more thoroughly discussed on p. 765.
Test Results and Clinical Significance
Increased Levels
These are acute streptococcal infections that will not immediately be associated with serologic changes because of the immunologic latency response.
Recent information suggests that rheumatic fever is associated with infection by rheumatogenic serotypes (M1, M3, M5, M6, M18, and M19), while glomerulonephritis follows infection by nephritogenic serotypes (M2, M12, M49, M57, M59, and M60). Streptococcal pyoderma is often associated with a reduced immunologic response as compared to throat infections.
Related Test
Throat culture (p. 765). The pharynx is a common anatomic site of streptococcal group A infection. Throat culture or rapid antigen testing are adequate to instigate antistreptococcal antibiotic therapy.
Syphilis Detection (Serologic Test for Syphilis [STS], Venereal Disease Research Laboratory [VDRL], Rapid Plasma Reagin [RPR], Fluorescent Treponemal Antibody [FTA])
Indications
These serologic tests are used to diagnose and to document successful therapy of syphilis.
Test Explanation
Syphilis is caused by the spirochete Treponema pallidum. The disease is divided into four stages: acute, secondary, latent, and tertiary. The acute stage is marked by the development of a chancre on the skin near the infection (usually the genitalia). The chancre develops about 3 to 6 weeks after inoculation and lasts for about 4 to 6 weeks. The secondary stage is highlighted by a rash (often on the soles and palms) and generalized lymphadenopathy. This stage lasts for about 3 months. The latent stage represents a period of disease inactivity and can last for 5 years. Some patients are cured of the infection during this stage. Many go into the tertiary stage marked by central nervous system (CNS), cardiovascular, and ocular signs and symptoms.
The immunologic tests for syphilis detect antibodies to T. pallidum. There are two groups of antibodies. The first group of tests detects the presence of a nontreponemal antibody called reagin, which reacts to phospholipids in the body (which are probably similar to lipids in the membrane of T. pallidum). The second group of tests detects antibodies directed against the Treponema organism itself. The nontreponemal antibody tests are grouped as serologic screening tests for syphilis and are relatively nonspecific. These antibodies are most often detected by the Wassermann test or the Venereal Disease Research Laboratory (VDRL) test. A more sensitive nontreponemal test is the Rapid Plasma Reagin (RPR) test. The VDRL and RPR tests, by virtue of their testing for a nonspecific antibody, have a high false-positive (or cross-reactive) rate. The VDRL test becomes positive approximately 2 weeks after the patient's inoculation with Treponema and returns to normal after adequate treatment. The test is positive in nearly all primary and secondary stages of syphilis and in two thirds of patients with tertiary syphilis.
If the VDRL or RPR test is positive, the diagnosis must be confirmed by the more specific Treponema test, such as the fluorescent treponemal antibody absorption test (FTA-ABS). The FTA test, which reacts to a specific treponemal antibody, is more accurate than the VDRL and RPR tests and becomes positive about 4 to 6 weeks after inoculation. To improve its specificity, non-pallidum antibodies are absorbed out of the patient's serum before the serum is added to the T. pallidum–impregnated slide. Anti–gamma globulin antibodies that are fluorescent are then added to the slide. If anti–T. pallidum antibodies exist in the patient's serum, they will react to the T. pallidum on the slide and the fluorescent anti–gamma globulin antibodies that were subsequently added will attach to the patient's serum antibody–T. palladium complex and make it visible under the fluorescent microscope. The FTA test is required before the diagnosis of syphilis can be made with certainty. A microhemagglutination test (MHA-TP) is also available and is comparable in accuracy to the “standard criterion” FTA-ABS test. Enzyme-linked immunosorbent assay (ELISA or EIA) methods are also available for detection of anti-treponemal/antibodies (IgG or IgM).
If the VDRL or RPR test is positive and the FTA-ABS is negative, other diseases that can cause positive results on screening serologic syphilis tests must be sought (Box 2-19).
Screening for syphilis is usually done during the first prenatal checkup of pregnant women using the VDRL or RPR test. Syphilis, if untreated, may cause abortion, stillbirth, or premature labor. The effect on the fetus can be CNS damage, hearing loss, or possible death. In patients who have symptoms compatible with primary syphilis, an FTA-ABS test is recommended. Congenital syphilis is difficult to distinguish from the passive immunity provided by a mother who has syphilis. However, in congenital syphilis the FTA-ABS result of the infant is usually much higher than the mother's. The term TORCH (toxoplasmosis, other, rubella, cytomegalovirus, herpes) has been applied to infections with recognized detrimental effects on the fetus. The effects on the fetus may be direct or indirect (e.g., precipitating abortion, premature labor). Included in the category of “other” are infections (e.g., syphilis). All of these tests are discussed separately.
During early primary syphilis, the first antibodies to appear are IgM, with IgG antibodies reaching significant titers later in the primary phase. As the disease progresses into the secondary phase, IgG Treponema pallidum antibodies reach peak titers. Treponema pallidum IgG antibodies persist indefinitely regardless of the course of the disease. If syphilis IgG and/or IgM is positive, results can be confirmed with FTA or MHA testing. The IgG- and IgM-specific antibodies assist in determining the etiology of neonatal syphilis. IgM does not pass through the placenta and if positive indicates active neonatal infection.
In general, serologic tests return to normal after successful treatment for syphilis. The earlier the disease is treated, the sooner the serologic tests return to normal. In the early primary stage the serologic tests may become negative in 2 to 4 months after successful antibiotic treatment. It may take longer than 1 year for the patient to convert to a seronegative result when treating later stages of the disease. In the tertiary stage the patient may never convert to negative. Testing should be routinely performed to document successful therapy.
Interfering Factors
• Excessive hemolysis and gross lipemia may cause false-positive STS test results.
• Excess chyle in the blood may cause false-positive STS test results. Testing should be performed after at least an 8-hour fast.
• Recent ingestion of alcohol may cause false-positive STS test results. Alcohol should be avoided for 24 hours before testing.
• Many conditions cause false-positive results when VDRL and RPR tests are used (see Box 2-19).
If the patient is tested too soon after inoculation and before antibodies have developed, the tests may be falsely negative. The test should be repeated in 2 months or the patient should be treated despite the negative test results if clinical suspicion is high.
Clinical Priorities
• If the patient is tested too soon after inoculation and before antibodies have developed, the tests may be falsely negative.
• Because the FTA tests for a specific treponemal antibody, it is more accurate than the VDRL and RPR tests. The FTA test becomes positive about 4 to 6 weeks after inoculation.
• Screening for syphilis is usually performed during the first prenatal checkup of pregnant women using the VDRL test.
Procedure and Patient Care
After
• Apply pressure or a pressure dressing to the venipuncture site.
• Check the venipuncture site for bleeding.
If the test is positive, instruct the patient to inform recent sexual contacts so they can be evaluated.
If the test is positive, be sure the patient receives the appropriate antibiotic therapy.
• If a screening serology test is positive, a more specific antitreponemal test is required to make the diagnosis.
Testosterone (Total Testosterone Serum Level)
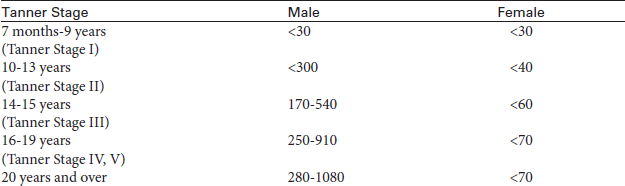
Indications
Testosterone levels are used to evaluate ambiguous sex characteristics, precocious puberty, virilizing syndromes in the female, and infertility or impotency in the male. This test can also be used as a tumor marker for rare tumors of the ovary and testicle.
Test Explanation
Androgens include dehydroepiandrosterone (DHEA) (p. 29), androstenedione, and testosterone. In the adrenal glands, DHEA is produced in the process of making cortisol and aldosterone. DHEA is also produced de novo by the testes or the ovaries. DHEA is the precursor of androstenedione, which is the precursor of testosterone (and estrogen).
Testosterone levels vary by stage of maturity (indicated by Tanner Stage). Serum concentrations of testosterone in both sexes during the first week of life average about 25 ng/dL. In male infants, values increase sharply in the second week to a maximum (mean about 175 ng/dL) at about 2 months, which lasts until about 6 months of age. In female infants, values decrease in the first week and remain low throughout early childhood. Levels increase during puberty to adult values.
In the male most of the testosterone is made by the Leydig cells in the testicle; this accounts for 95% of the circulating testosterone in men. In the female about half of the testosterone is made by the conversion of DHEA to testosterone in the peripheral fat tissue. Another 30% is made by the same conversion of DHEA in the adrenal gland, and 20% is made directly by the ovaries.
Approximately 60% of circulating testosterone binds strongly to sex hormone–binding globulin (SHBG), which is also called testosterone-binding globulin. Most of the remaining testosterone is bound loosely to albumin, and approximately 2% is free or unbound. The unbound portion is the active component. Most assays for testosterone measure the total testosterone (i.e., bound and unbound portions). The free testosterone can be measured where the testosterone binding proteins may be altered (obesity, cirrhosis, thyroid disorders). Free testosterone is estimated in this panel by an indirect method, equilibrium ultrafiltration. It can be reported as a percentage of total testosterone, or as an absolute number.
In males a biofeedback mechanism exists that starts in the hypothalamus. Gonadotropin-releasing hormone (GnRH) induces the pituitary to produce luteinizing hormone (LH) (called interstitial cell–stimulating hormone in the male) and follicle-stimulating hormone (FSH). LH stimulates the Leydig cells to produce testosterone. FSH stimulates the Sertoli cells to produce sperm. Testosterone then acts to inhibit further secretion of GnRH.
Physiologically, testosterone stimulates spermatogenesis and influences the development of male secondary sexual characteristics. Overproduction of this hormone in the young male may cause precocious puberty. This can be caused by testicular, adrenal, or pituitary tumors. Overproduction of this hormone in females causes masculinization, which is manifested as amenorrhea and excessive growth of body hair (hirsutism). Ovarian and adrenal tumors/hyperplasia and medications (e.g., danazol) are all potential causes of masculinization in the female. Reduced levels of testosterone in the male suggest hypogonadism or Klinefelter syndrome.
Dihydrotestosterone (DHT) is the principal androgen made in body tissues, particularly the prostate. Levels of DHT remain normal with aging, despite a decrease in the plasma testosterone, and are not elevated in benign prostatic hyperplasia. Measurement of this hormone is useful in monitoring patients receiving 5 alpha-reductase inhibitor therapy such as finasteride or chemotherapy, which may affect prostate function. It is also useful in evaluating patients with possible 5 alpha-reductase deficiency.
There are several testosterone stimulation tests that can be performed to more accurately evaluate hypogonadism. Human chorionic gonadotropin, clomiphene, and GnRH can be used to stimulate testosterone secretion.
17-ketosteroids (17-KS) are metabolites of the testosterone and non-testosterone androgenic sex hormones that are excreted in the urine.
Methods used for the measurement of testosterone include radioimmunoassay and extraction chromatography. There is a slight diurnal variation in the secretion of testosterone. Levels are maximal around 7 AM and minimal around 8 PM.
Interfering Factors
Drugs that may cause increased testosterone levels include anticonvulsants, barbiturates, estrogens, and oral contraceptives.
Drugs that may cause decreased testosterone levels include alcohol, androgens, dexamethasone, diethylstilbestrol, digoxin, ketoconazole, phenothiazine, spironolactone, and steroids.
Test Results and Clinical Significance
Increased Levels (Male)
Idiopathic sexual precocity: This is usually because of oversecretion of LH, which stimulates the testicles to produce testosterone.
Pinealoma: This is a hypothalamic tumor that produces an increased quantity of GnRH, which stimulates the pituitary to produce LH, which in turn stimulates the testicles to produce testosterone.
Encephalitis: This viral infection of the CNS can stimulate the hypothalamus to produce an increased quantity of GnRH, which stimulates the pituitary to produce LH, which in turn stimulates the testicles to produce testosterone.
Congenital adrenal hyperplasia: An enzyme deficiency in the production of cortisol causes an accumulation of large amounts of DHEA. DHEA is a precursor of androstenedione, which is a precursor of testosterone.
Adrenocortical tumor: Neoplasm involving the adrenal gland can produce large amounts of testosterone or DHEA. DHEA is a precursor of androstenedione, which is a precursor of testosterone.
Testicular or extragonadal tumor: Leydig cell tumors can produce testosterone, which can cause precocious puberty in males. However, no spermatogenesis occurs because gonadotropin hormones are not produced and are, in fact, inhibited.
Hyperthyroidism: These patients have elevated bound testosterone because of elevated SHBG proteins. This causes elevation of the total testosterone levels.
Testosterone resistance syndromes: These patients resist the effect of testosterone on tissue. In response, higher levels of testosterone are secreted.
Decreased Levels (Male)
Klinefelter syndrome: These patients have an extra X chromosome (XXY). This syndrome is associated with primary testicular failure.
Cryptorchidism: These patients usually have normal testosterone levels, but occasionally testicles that fail to descend into the scrotum can be atrophic.
Primary and secondary hypogonadism: Infection, tumor, or congenital abnormalities are all possible causes of primary (testicular) or secondary (pituitary) failure.
Trisomy 21: The pathophysiology of this genetic defect is not defined.
Orchiectomy: The testicles must both be removed. Surgical removal of just one testicle does not cause deficient testosterone levels.
Hepatic cirrhosis: These patients have reduced proteins and therefore have reduced amounts of bound testosterone, which makes up most of the total testosterone that is measured.
Increased Levels (Female)
Ovarian tumor: Arrhenoblastoma is an uncommon ovarian tumor that can produce testosterone.
Adrenal tumor: Neoplasms involving the adrenal gland can produce large amounts of testosterone or DHEA. DHEA is a precursor of androstenedione, which is a precursor of testosterone. Hirsutism in females is common with these tumors.
Congenital adrenocortical hyperplasia: An enzyme deficiency in the production of cortisol causes an accumulation of large amounts of DHEA. DHEA is a precursor of androstenedione, which is a precursor of testosterone. In females this can result in pseudohermaphroditism (ambiguous genitalia).
Trophoblastic tumor: These tumors (hydatidiform mole, choriocarcinoma) produce hCG, which can stimulate the production of testosterone.
Polycystic ovaries: This syndrome is associated with obesity, hirsutism, and amenorrhea. Patients have increased testosterone levels. The pathophysiology is not well defined.
Idiopathic hirsutism: The pathophysiology of this observation is not known.
Related Test
Adrenal Steroid Precursors (p. 29). Androstenedione is used in the diagnosis of virilizing syndromes, especially in the female.
Thromboelastography (Thromboelastometry)
Test Explanation
Hemostasis is a well-regulated process in which the blood forms localized clots when the integrity of the vascular system is breeched. Trauma, infection, and inflammation all activate the blood's clotting system, which depends on the interaction of two separate systems: enzymatic proteins (clotting factors, intrinsic and extrinsic systems [Figure 2-29]) and platelets. The two systems work in concert to plug defects in the broken vessels. The clots that form in this process need to be of sufficient strength to resist dislodgement. If a particular clotting factor is dysfunctional or absent, as in hemophilia, an insufficient amount of fibrin forms. Similarly, massive consumption of clotting factors in a trauma situation decreases the amount of fibrin formed. Inadequate numbers of platelets resulting from trauma, surgery, or chemotherapy also decrease platelet aggregation, as do genetic disorders, uremia, or medication therapy. Ultimately, reduced fibrin formation or platelet aggregation results in clots of inadequate tensile strength. This hypocoagulable state is associated with excessive bleeding. Conversely endothelial injury, stasis, cancer, genetic diseases, or other hypercoagulable states lead to thrombosis formation causing thromboembolic events.
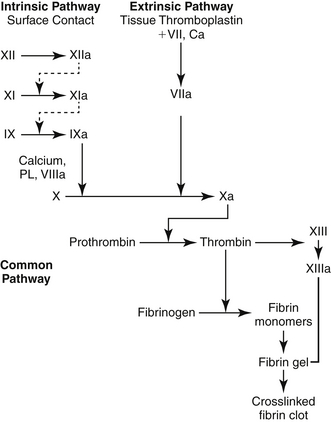
In this test whole blood is rapidly transferred to a cuvette. Clot-activating catalysts may be added to speed up the process. The cuvette is rotated as a clot forms in a machine. The fibrin in the clot creates fluid resistance that is determined by a sensor transducer (also placed within the cuvette) that converts the tensile strength of the client to an electronic signal displayed on a graph. Thrombosis time and lysis time can be calculated and is reported as a coagulation index. This test is able to represent all global analysis of the hemostatic function from initial thrombin generation to clot lysis. When plotted on a graph, specific patterns can be identified, including normal, hemophilia, thrombocytopenia, decreased platelet function, increased fibrinolysis, and hypercoagulation.
This test is used to identify patients who are hypercoagulable and may experience a thromboembolic phenomenon when immobile (e.g., after surgery or trauma). It is particularly helpful in cardiac surgery and liver transplantation. It is also used to determine hyperfibrinolysis. Finally this shows the complete evaluation of platelet function. Usually three separate tracings using different reagents can determine the percent of platelet inhibition instigated by heparin, aspirin, and antiplatelet drugs (Plavix, Ticlid). This test correlates better with operative bleeding than does bleeding time, closure time (p. 404), or thromboxane levels. With the present instrumentation, point of service (e.g., in the operating room) testing can be performed.
Procedure and Patient Care
Related Tests
Platelet Aggregation (p. 398). This is a measure of the abilities of the platelets to aggregate.
Platelet Function Assay (p. 404). This is a measure of platelet function.
Platelet Count (p. 401). This is an account of the number of thromboses.
Coagulating Factor Concentration (p. 163). These tests measure the quantity of each specific factor.
Factor V-Leiden (p. 231). This is an inherited abnormality affecting the coagulation cascade.
Protein C, Protein S (p. 432). These are important inhibitors of the coagulation system.
Plasminogen (p. 394). This protein is involved in the fibrinolytic process.